
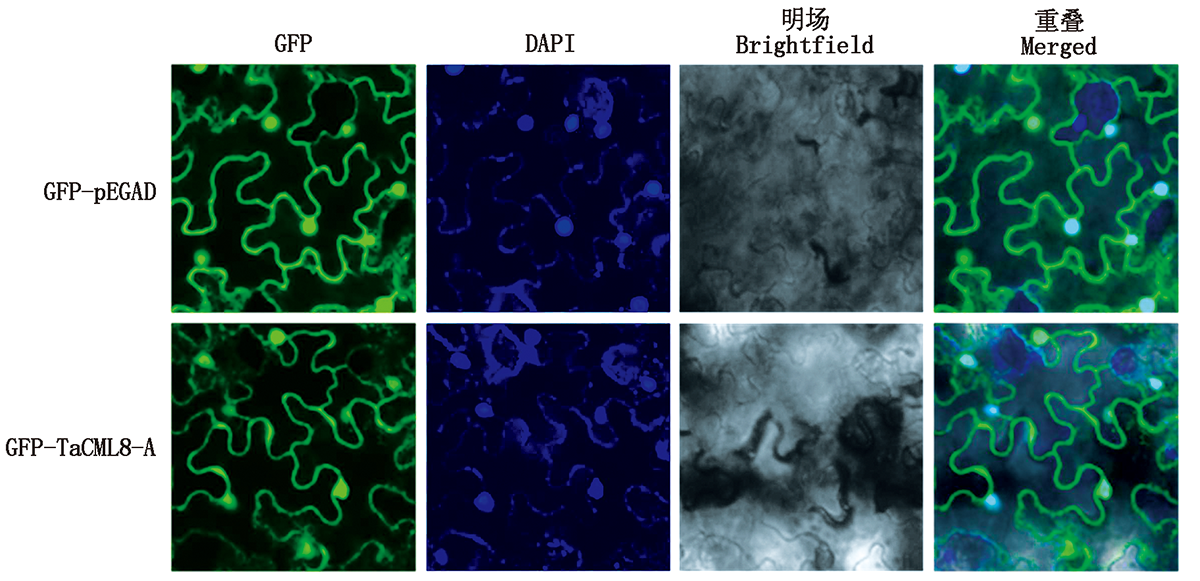
In order to explore stress resistance genes in wheat and study the molecular mechanism of calmodulin-like genes in plant stress resistance,a calmodulin-like gene TaCML8-A was cloned from wheat by electronic cloning combined with RT-PCR.Bioinformatics analysis showed that the open reading frame length of the gene was 519 bp,encoding 172 amino acid sequences,including PTZ00184 and FRQ1 superfamily,four EF-hand domains,including four calcium binding sites.The molecular weight of the encoded protein was 18.31 ku and the isoelectric point was 4.54.It belonged to acidic protein.Subcellular localization showed that TaCML8-A protein was distributed in the nucleus and cell membrane.Nucleotide sequence alignment showed that TaCML8-A had the closest genetic relationship with rice OsCML14,with a similarity of 79.17%.qRT-PCR results showed that the expression of TaCML8-A gene in shoot and root of wheat increased under salt,osmotic and cold stress,indicating that plants might respond to thoese stresses by increasing the expression of TaCML8-A.During heat shock,TaCML8-A gene was induced and inhibited in root and shoot,respectively,so as to play different functions.The calmodulin-like gene TaCML8-A was successfully cloned from wheat and analyzed for expression.It is preliminarily speculated that the gene might be involved in regulating abiotic stress of plants,so as to provide basic theoretical support for the follow-up study of its biological function.
The objective of this work was to develop novel,convenient,and reliable molecular marker in soybean.Twelve soybean germplasm accessions from different geography region were used to develop large-size InDel markers based on 10 depth genomic re-sequencing.Developed InDel markers were used to construct DNA fingerprint of 96 varieties in Huanghuaihai Uniform Test in 2018.The results showed that a total of 66 561 InDel markers,with insertion/deletion fragment size larger than 20 bp,were discovered among the 12 accessions.The distribution pattern among the genome of these InDel markers were also illustrated.The ratio of the InDel in intron,upstream of the gene,downstream of the gene,intergenic region,intragenic region,5'-UTR and 3'-UTR were 12.35%,25.83%,20.44%,34.39%,0.19%,0.93%,1.51%,respectively.There were 42 453 InDels yield insertion/deletion fragment size between 20 to 40 bp,13 044 InDels between 41—60 bp,5 034 InDels between 61—80 bp,2 285 InDels between 81—100 bp,and 2 413 InDels larger than 100 bp.Then,160 InDel markers were randomly selected according to the insertion/deletion fragment size throughout the whole genome.As results,thirty-two InDel markers,that only yield 2 alleles in for each locus and could be distinguished easily on agarose gel,were developed and validated through primer design,PCR and agarose gel electrophoresis.Additionally,DNA fingerprint of 96 varieties in Huanghuaihai Uniform Test in 2018,were constructed using the 32 developed InDel markers.As results,the average purity of the tested soybean varieties was 96.84%,and no varieties shared the same name.The InDel markers developed in this study were stable,reliable and user friendly.They could be used to construct DNA fingerprint and test seed purity of soybean.
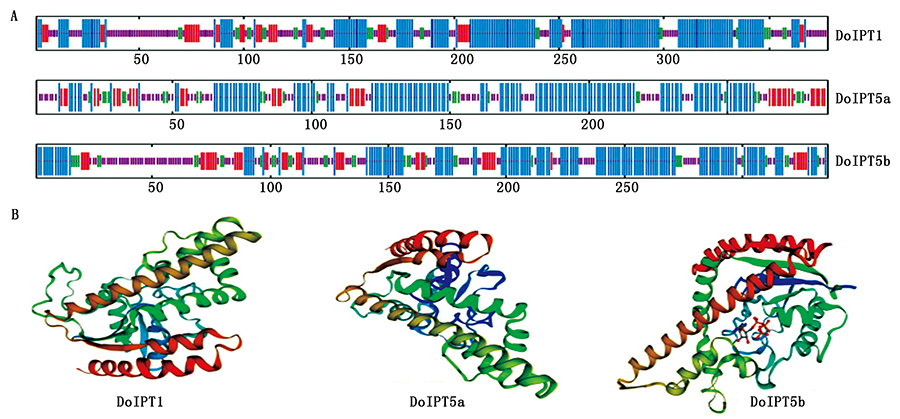
The aim of this research was to clone IPT(Isopentenyl transferase,IPT ) genes in yam,predict the structure and property of DoIPT proteins and identify the expression of DoIPTs during bulbil sprouting.DoIPTs were cloned by homologous cloning techniques.The structure and property of DoIPT proteins were predicted by bioinformatics analysis.Real-time PCR technique was applied to identify the expression of these genes.Three nucleic acid sequences of 1 398,1 044,1 349 bp were obtained from yam.The three sequences were named DoIPT1,DoIPT5a and DoIPT5b,and the registration numbers of these genes were MW353160,MW353016 and MW353017,respectively.The reading frame lengths were 1 137,861,1 011 bp respectively,and the encoded amino acid lengths were 378,286,336 aa,respectively.Bioinformatics analysis showed that DoIPTs were unstable,hydrophilic and external family proteins.There were two signal peptides in DoIPT5b,and no signal peptide was found in the other two proteins.The secondary structures in the three proteins were consistent with the prediction models of the tertiary structures of three genes.The encoded regions of DoIPTs had high similarity with the isopentenyl transferase protein of other plants.Coding areas of DoIPTs showed that the N-terminal regions of DoIPTs contained the P-loop NTPase structural domain GXXGXGKS(T),which was able to catalyse cytokinin synthesis.The results of Real-time PCR technique showed that the expression of DoIPT5a in sprouting bulbils was highest under the condition of water shortage at 22 ℃;and the expression of DoIPT1 and DoIPT5b genes was the highest in a humid environment;the expression of DOIPTs genes raised with paclobutrazol treatment.These results laid a foundation for further study on the function of DoIPTs in yam.
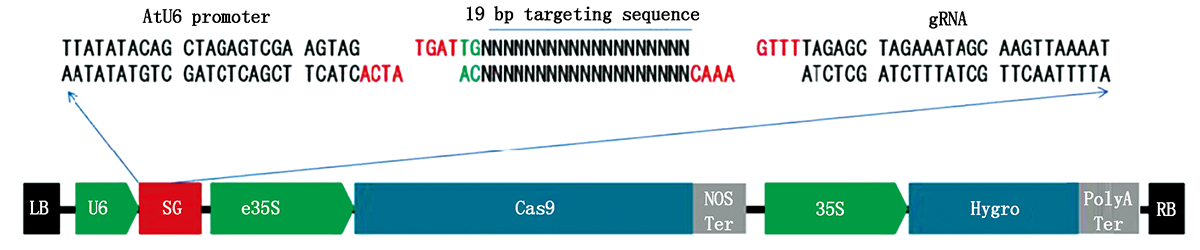
Tomato belongs to the cold sensitive model plant,which is vulnerable to chilling injury during its growth,thus affecting its yield.In order to provide a theoretical basis for further revealing the molecular mechanism of cold resistance of tomato plants and breeding tomato cold resistant varieties,tomato germplasm resource No.25 of vegetable genetics,Breeding and Biotechnology Laboratory was used as material,and tomato transcription factor SlMYB-related 2 was used as research object.Based on CRISPR/Cas9 gene knockout vector,tomato positive plants were obtained by Agrobacterium transformation.The VIGS transient silencing expression vector was constructed,and the wild-type and and virus transformed plants without inserted target gene fragment were used as control.The three groups of plants were treated with low temperature at 4 ℃(16 h day / 8 h night,60% humidity).The contents of physiological indexes related to cold resistance were measured and its cold tolerance was compared.The results showed that CRISPR/Cas9 gene knockout vector was constructed and two CRISPR silenced positive plants were obtained.Constructed the expression vector of VIGS silence,after low temperature treatment,it was found that with the extension of low temperature treatment time,the growth trend of proline and soluble sugar in SlMYB-related 2 gene transformation group was slower than that in control tomato group(wild type carrying TRV,CK)and wild group(WT),and the content was lower than that in CK group and wild group.However,the content of malondialdehyde was higher than that of CK group and wild group,which proved that the cold resistance of tomato plants with transient expression of VIGS was significantly lower than that of CK group and wild group.Through the analysis of cold resistance of tomato plants after gene transient silencing expression,it was found that SlMYB-related 2 gene played a positive regulatory role in low temperature stress.
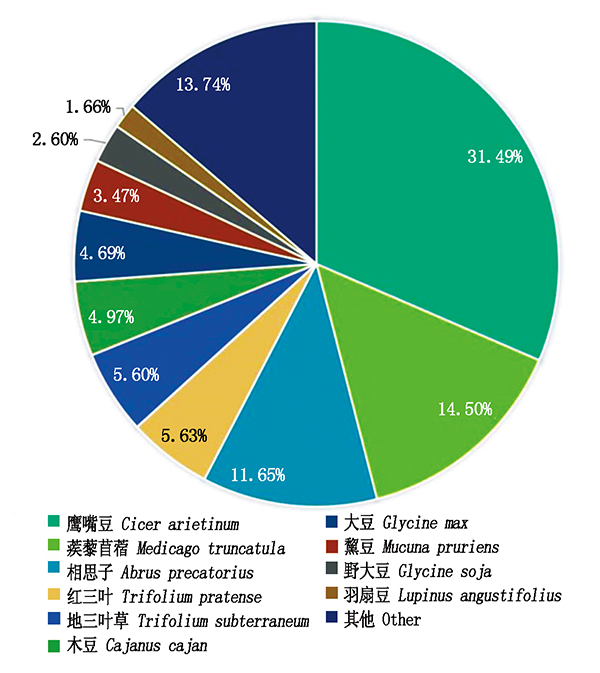
In order to obtain transcriptome information and functional genes expression characteristics of Astragalus complanatus,the leaves of A.complanatus were used as material,the high-throughput transcriptome sequencing and bioinformatics analysis were performed using Illumina HiSeq platform.A total of 19 280 Unigenes were assembled with total sequence length of 23 472 470 bp,of which the GC content was 42.74%,indicating a high quality of sequencing and assembly.Blast analysis showed 12 541,10 120,9 412,8 953,7 494,5 052 Unigenes got annotation in Nr,Swiss-Prot,GO,KEGG,KOG and COG databases,respectively.The annotated Unigenes of A.complanatus were mainly homologuous to leguminous plants,in particular,highly matched to the subfamily Pteriformis plant Cicer arietinum,Medicago truncatula and Abrus precatorius with a respective percentage of 31.49%,14.50% and 11.65%.Annotated Unigenes were divided into 3 GO classifications,354 KEGG metabolic pathways and 25 KOG function categories.The most abundant categories in each database were metabolic process,general function prediction only and purine metabolism,respectively.These results showed that the leaf cells of Astragalus membranaceus had active metabolism and abundant gene expression in the seedling stage.Additionally,a lot of Unigenes were annotated to the infectious disease category in KEGG database,suggesting a potential medicinal value of the plant part of Astragalus membranaceus.A total of 5 849 simple sequence repeats(SSRs)were identified by MISA software,with an occurrence frequency of 30.34%.The SSRs in the genome of Astragalus membranaceus were abundant and diversified,containing single base to six base repeats,among which the mono-nucleotide SSRs had the largest number with a frequency of 40.56%.The main types of SSR motifs involved A/T,AG/CT and AAG/CTT.
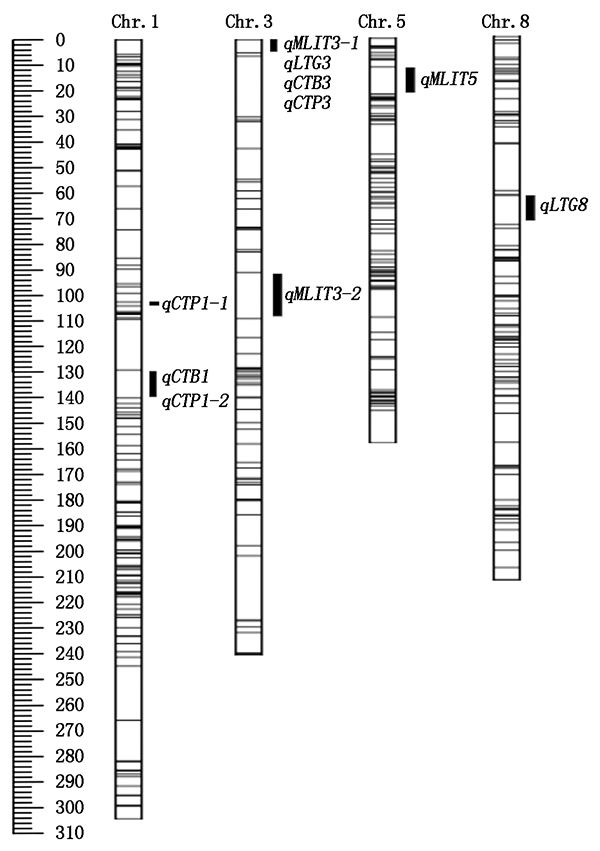
To map QTLs that control cold tolerance at germination and bud stages,to explore the genetic mechanism,a recombinant inbred line(RIL)population was used as experimental material.The RIL population contained 189 lines derived from the cross between Caidao,a japonica rice variety that had strong cold tolerance and was adopted as the female parent,and WD,a indica rice variety that had weak cold tolerance and was used as the male parent.The QTLs that control cold tolerance at germination and bud stage were mapped between four phenotypic indexes and 978 Bin markers by IciMapping 4.2.The four phenotypic indexes included relative germination rate,average germination days,seedling rate and cold tolerance level.The results showed that two pairs of epistatic QTLs related to cold tolerance at bud stage were mapped.The LOD scores were 5.70 and 5.55 and the phenotypic contribution rates were 13.22% and 39.87%,respectively.In addition,five and five QTLs that were related to cold tolerance at germination and bud stage were mapped,respectively.These QTLs were located on chromosomes 1,3,5 and 8.The LOD score and phenotypic contribution rate of these QTLs ranged from 2.55 to 7.07 and from 6.41% to 17.17%,respectively.qLTG3 had the highest phenotypic contribution rate among the five germination stage QTLs and was also related to cold tolerance at germination stage.qCTP1-2 had the maximum of phenotypic contribution rate among the five bud stage QTLs.qCTB1,qCTP1-2 and qLTG8 had not been reported.The QTLs and epistatic QTLs detected will provide a reference for the genetic research and improvement of cold tolerance in rice.
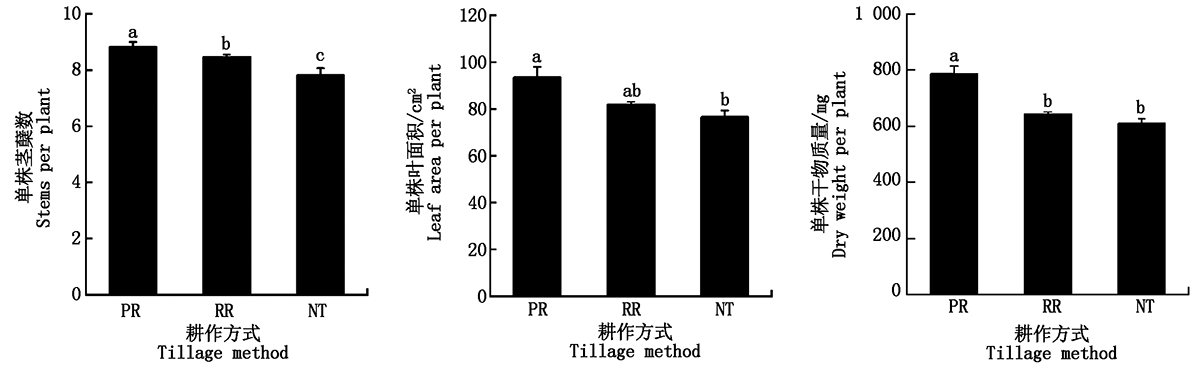
To investigate the cultivation approach achieving robust seedlings and high yields of wheat in rice stubble,the effects of three tillage methods,including plow tillage followed by rotary tillage(PR),rotary tillage twice(RR),and no-tillage(NT),on the growth and physiology of main stem and tillers of wheat seedlings and their single spike yield at maturity were studied under the conditions of rice straw retaining with full amount.The results showed that PR increased the stems number per plant at the beginning of over-wintering by 1.5%,12.8%,respectively,compared with RR and NT,with significant differences(P<0.05).Leaf area and dry matter weight per plant under PR were 94 cm2 and 787 mg,respectively,which were significantly higher than those under NT(P<0.05).Compared with NT,PR significantly improved activities of nitrate reductase(NR),glutamine synthase(GS),and glutamate synthase(GOGAT)of the top full expanding leaves in the main stem and the first,second,and third tillers(P<0.05).PR also significantly promoted the activities of diphosphate carboxylase(Rubisco),pyruvate phosphate dikinase(PPDK),triosephosphate isomerase(TPI),sucrose synthase(SS-Ⅱ),and sucrose phosphate synthase(SPS),and enhanced SPAD and soluble sugar content(P<0.05).And compared with NT,PR significantly increased the nitrogen content in seedling leaves of main stem,second and third tillers(P<0.05).Moreover,PR and RR increased leaf area and dry matter weight of the main stem and the first and second tillers.Therefore,compared with RR and NT,PR was more beneficial to the synergistic improvement of nitrogen metabolism,glucose metabolism,and light energy utilization at the seedling stage,increasing photosynthetic productivity and forming robust seedlings.The improvement of carbon and nitrogen metabolism and photosynthetic capacity by RR was higher than that by NT but was weaker than that by PR.There were no significant differences between PR and RR in the grain number per spike,single grain weight of the main stem(P>0.05),but they were significantly higher than those under NT(P<0.05).The grain number per spike in the first and second tillers under PR was significantly higher than that under RR and NT(P<0.05),but single grain weight and grain yield per spike of the first,second,and third tillers were not significantly different between PR and RR(P>0.05).In conclusion,PR could improve the activity of glucose and nitrogen metabolism and photosynthetic physiology of the main stem and the first and second tillers of wheat in rice stubble,resulting in the increases of the grain number per spike and the productivity per spike through forming strong seedlings.
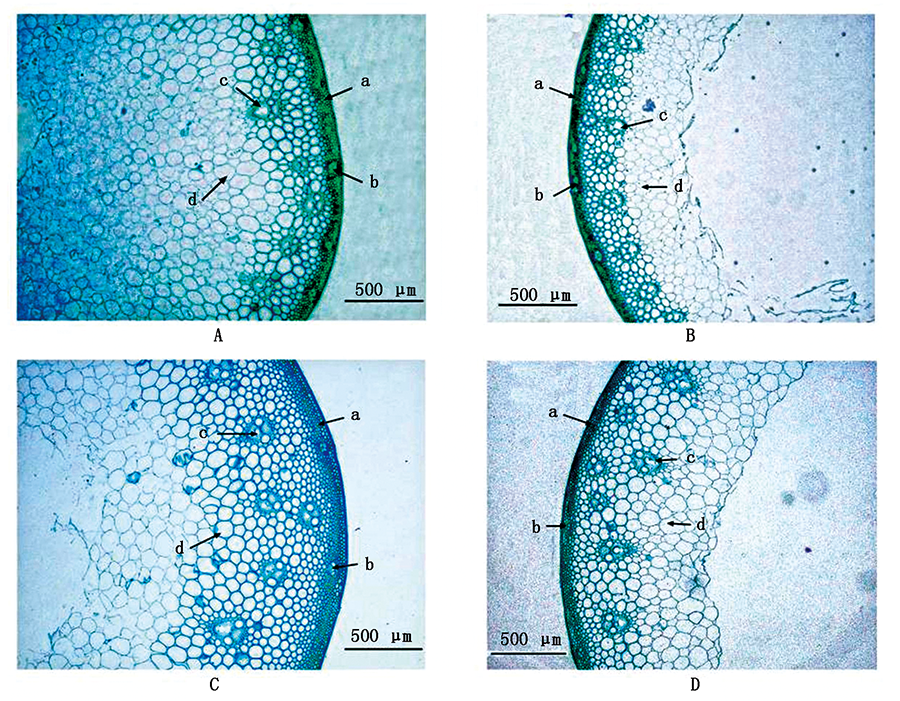
In order to explore the key factors affecting stem strength and screen the valuable parent resources for wheat "strong stem breeding" ,72 wheat varieties(lines)were used as materials.Firstly,the strength of stems in flowering stage,filling stage and milk stage of wheat was comprehensively analyzed and all the materials were grouped into clusters.On this basis,the genetic variation of lignin content,cellulose content and microstructure of wheat stem and their relation with stem strength were further analyzed,and the key characters related to stem strength were defined.In addition,principal component analysis(PCA)was used to extract the main factors of stem cross section microstructure,and calculate the score of comprehensive factors,and further cluster analysis was carried out combined with lignin and cellulose content,so as to comprehensively evaluate the performance of stem strength related traits of different wheat varieties(lines).The results showed that the mechanical tissue thickness decreased significantly or extremely significant at the same time as the strength of wheat stem weakened from flowering stage to filling stage.Moreover,parenchyma thickness,mechanical tissue thickness,large vascular bundle area,lignin and cellulose content in high stem strength wheat were much higher than those in low stem strength wheat.Correlation analysis also showed that mechanical tissue thickness,large vascular bundle area,lignin and cellulose content were significantly of extremely significantly correlated with stem strength.Finally,the lignin content,cellulose content and the main component comprehensive factor scores of the stem microstructure were used as three variables for cluster analysis,and a total of 18 wheat materials with outstanding performance in the stem microstructure and biochemical indicators were screened out.These wheat materials can be used as important genetic resources for wheat "strong stem breeding".
Using rice variety Shendao 9 as an entry,a field experiment was conducted in 2019—2020 to study the effect of nitrogen application pattern and row spacing on canopy structure and yield of rice under split-plot design.The main plot treatments were zero nitrogen(A0),farmer's pattern(A1),low basal nitrogen(A2),and basal nitrogen backward(A3).The subplot treatments were conventional method(row spacing of 30 cm,B1),reducing row spacing(row spacing of 25 cm,B2),and narrow-wide row(row spacing of 40 cm+20 cm,B3).The layer upon layer cut method was used to investigate the leaf area index(LAI),photosynthetic active radiation(PAR),photosynthetic characteristics of flag leaves,and grain yield of rice.The results showed that nitrogen application pattern and row spacing had extremely significant interaction effects on grain yield,the maximum value(9.85 t/ha)appeared in A3B2.Compared with A1 and A2,the effective spikelets per unit area,kernel-setting rate,net photosynthetic rate(Pn),stomatal conductance(Gs)and transpiration rate(Tr)of A3 increased by 4.31%—10.55%,2.87—4.09 percentage points,4.84%—9.12%,14.08%—15.71% and 11.33%—15.83%,respectively.Meanwhile,rice obtained higher interception rate of PAR,population leaf area index under A3.Compared with B1 and B3,the spikelets per unit area,and population leaf area index(LAI)of B2 increased by 7.57%—9.97% and 4.29%—20.43%,respectively.And interception rate of PAR of B2 was 88.99%.Given the results of grain yield,PAR,and photosynthetic characteristics in rice of two years,it could be concluded that the basal nitrogen backward pattern combined with 25 cm row spacing could enhance spikelets per unit area,kernel-setting rate,1000-grain weight,improve the population structure and obtain higher yield.
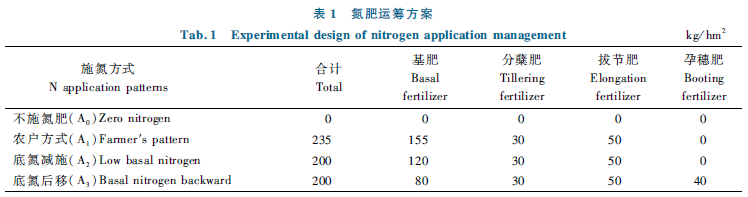
In order to explore the effects and mechanism of grain filling regulators on inferior spikelets filling of large panicle rice varieties,large panicle rice variety Jiaoyuanyou 216 was selected as material and an open field experiment was conducted by spraying exogenous grain filling regulators(Helifeng and Xinmeizhouxing)at heading stage.Meanwhile,endogenous hormone contents,expressions of filling-related miRNAs and their targets,and genes encoding filling-related proteins and sucrose-starch metabolism key enzymes were measured.The results should that the two grain filling regulators of Helifeng and Xinmeizhouxing significantly increased the yield of Jiaoyuanyou 216,the 1000-grain weight,filling rate and initial grain filling potential of inferior spikelets.Specifically,compared with the control(spraying water),the 1000-grain weight of inferior spikelets increased by 16.07% and 15.89%,respectively.Content of IAA in inferior spikelets increased significantly at 9,15,21 d after flowering,and the content of Z+ZR increased significantly at 9,21 d after flowering.Under Helifeng treatment,expressions of miR167a-c,miR167d-j and miR1432 were significantly down-regulated at 6 d after flowering,OsGF14b and OsGF14f were significantly down-regulated at 12 after flowering. What's more,expression of OsGLP3,a gene encoding germin-like protein was significantly up-regulated at 6 d after flowering,and expressions of sucrose-starch metabolism key enzyme related genes were significantly up-regulated at 6,12 d after flowering.However,under Xinmeizhouxing treatment,expressions of miR167a-c,miR167d-j,miR1432 were significantly down-regulated at 6 d after flowering,and expressions of OsGF14b and OsGF14f were significantly down-regulated at 12 d after flowering.More importantly,expressions of OsGLP3 and sucrose-starch metabolism key enzyme related genes were significantly up-regulated at 6,12 d after flowering.Correlation analysis results showed that the filling rate of inferior spikelets was significantly negatively correlated with the expression of miR1432 and OsGF14f,while was significantly positively correlated with the content of IAA.Therefore,Helifeng and Xinmeizhouxing may regulate the expressions of filling-related miRNAs and their targets,as well as the genes of filling-related proteins and sucrose-starch metabolism key enzymes in the pre-and mid-filling period and increase the content of IAA and Z+ZR to promote inferior spikelets filling.
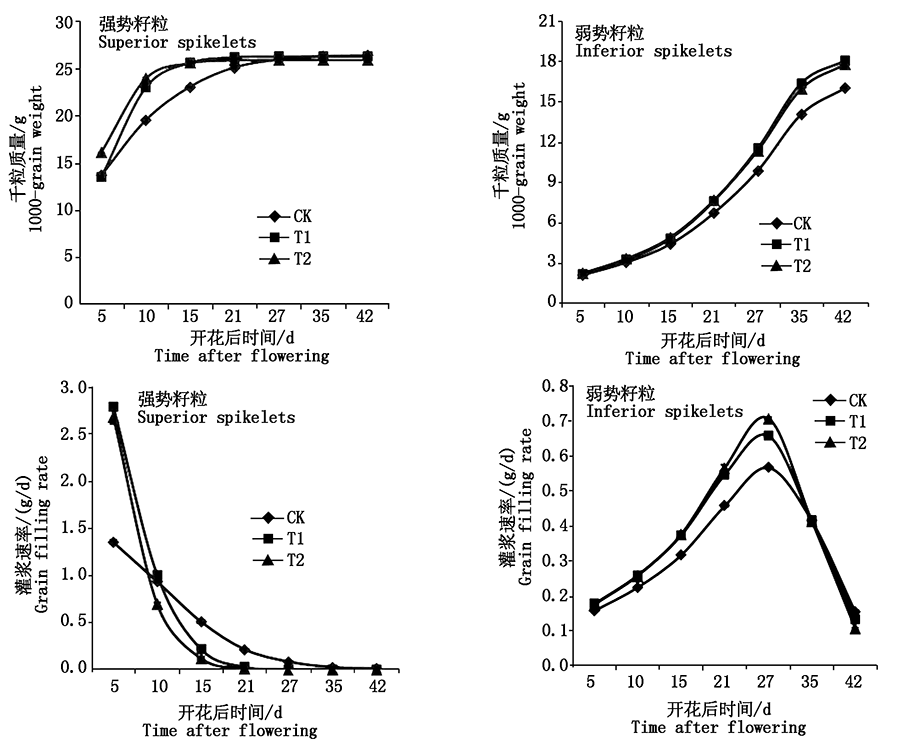
To study the effect of soybean rhizobia inoculation under different nitrogen levels on soybean growth and nodulation and nitrogen fixation,soybean Williams 82 was used as the test material to be planted by vermiculite method,and nitrate treatments with three concentrations of no nitrogen,low nitrogen and high nitrogen were set up.Inoculate USDA110 soybean rhizobia to study the effect of different nitrogen levels on soybean nodulation and nitrogen content.The results showed that different levels of nitrogen application had different effects on the number of nodules,nodule dry weight and root dry weight.The application of high-nitrogen nutrient solution could inhibit soybean nodulation,while no nitrogen treatment promoted soybean nodulation.At the same time,the root dry weight showed the opposite trend,the root dry mass of the no nitrogen treatment was the smallest,and the root dry mass of the high-nitrogen treatment was the largest.Under different nitrogen levels,the nitrogen content of different organs of soybean showed different changing trends between inoculation and non-inoculation.Under different nitrogen levels,the nitrogen content under high nitrogen treatment was higher than low nitrogen and no nitrogen treatment,and the nitrogen content of different organs showed the phenomenon of leaf>root>stem overall.Under different nitrogen levels,the chlorophyll values(SPAD)of different leaf positions showed that there was no significant difference between inoculation and non-inoculation.The measurement results of leaf morphological indicators showed that the leaf area,leaf circumference,leaf length and leaf width of high nitrogen treatment were significantly higher than low nitrogen and no nitrogen treatments.Leaf area and leaf circumference after nitrogen-free inoculation were significantly greater than those without nitrogen-free inoculation,indicating that inoculation not only promoted root growth,but also stimulated leaf enlargement.It can be seen that rhizobium inoculation had different changes for nodulation,nitrogen fixation and growth and development of soybean seedlings under different nitrogen levels.Appropriate increase in nitrogen nutrition combined with the use of rhizobia was an effective way to promote the growth and development of soybean and the utilization and absorption of nitrogen.
In order to explore the effects of different concentrations of exogenous melatonin on the growth of potato seedlings under low temperature stress after cold acclimation,using Yunnan main potato variety Hezuo 88 as material,the effects of different concentrations of exogenous melatonin(50,100,150 μmol/L)on the growth and active oxygen metabolism system of potato seedlings under low temperature stress(4 ℃ 14 d ,-2 ℃ 12 h)were studied.The purpose of this study was to explore the feasibility of exogenous MT in alleviating the injury of potato seedlings caused by low temperature stress.The results showed that compared with potato seedlings under low temperature stress,the survival rate of seedlings treated with 50 μmol/L MT increased by 38.89 percentage point,and the plant height and stem diameter increased by 2.92% and 1.86%,the contents of SS,SP and Pro increased by 12.66%,13.80% and 1.96%,the activities of POD and CAT increased by 29.24% and 351.62%.The plant height and stem diameter of seedlings treated with 100 μmol/L MT increased by 16.97% and 2.82%,the contents of MDA and H2O2 decreased by 19.35% and 0.48%,the activities of POD and CAT increased by 55.44% and 213.56%.The survival rate of seedlings treated with 150 μmol/L MT increased by 27.78 percentage point,the plant height and stem diameter increased by 5.62% and 13.20%,the content of MDA decreased by 9.45%,the content of Pro increased by 38.70%,SOD,POD and CAT activity increased by 52.61%,25.25% and 300.94%,and the content of AsA and GSH increased by 3.48% and 3.97%.The results showed that exogenous MT could promote the growth of potato seedlings,reduce the content of MDA and H2O2 in leaves,promote the accumulation of SS,SP and Pro,increase the activities of SOD,POD and CAT,and increase the content of AsA and GSH.Thus,it can alleviate the damage of low temperature stress to potato seedlings,and then improve the ability of potato seedlings to resist low temperature stress.In summary,50 μmol/L MT treatment could better improve the stress resistance of potato seedlings under low temperature stress.

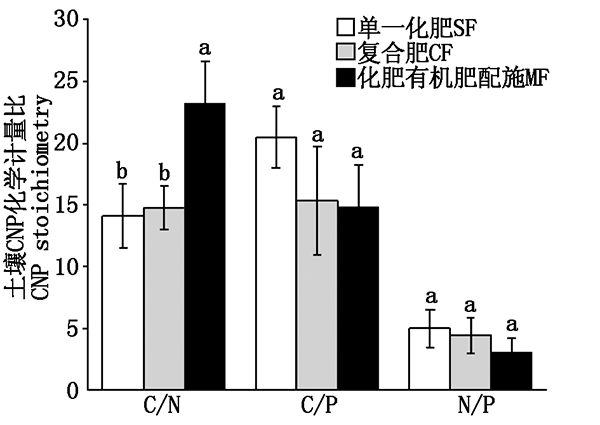
Stoichiometry characteristics of soil CNP and MBC,MBN,MBP,and the changes of enzyme activity with different fertilization can help to understand the relationship between fertilization and soil nutrient balance,and thus formulate more reasonable fertilization measures.Published literature data on the changes of CNP status and enzyme activity in fertilized soil were collected,and the changes of CNP stoichiometry and enzyme activity in cultivated soil nutrient/biomass with different fertilization were analyzed by Meta-analysis.The results showed that the C/N under single fertilizer,compound fertilizer and the application of chemical fertilizer and manure was 14.1,14.8 and 23.2,the value of C/P was 20.4,15.3 and 14.8,the value of N/P was 5.03,4.43 and 3.10,respectively.There was no significant difference in MBC/MBN and MBN/MBP with different fertilization.The ratio of MBC/MBP was:application of fertilizer and manure>single fertilizer>compound fertilizer.Different fertilizations could improve the activities of soil sucrase,phosphatase,urease and catalase,especially the application of chemical fertilizer and manure(which increased urease activity by 100%).It showed that the application of chemical fertilizer and manure could significantly affected the stoichiometric ratio of CNP in soil,enhanced microbial activities and promoted microorganisms to release more enzymes conducive to soil nutrient mineralization.It was an effective way to improve crop yield and ensure soil health in agricultural production.Soil enzyme activity was restricted by soil fertility and microbial process.A certain correlation between CNP stoichiometric ratio and soil enzyme activity was observed,which was also affected by land use type,soil pH and rotation system.The study of soil nutrient-microorganism-enzyme activity system can provide a theoretical basis for optimizing fertilization management in farmland system.
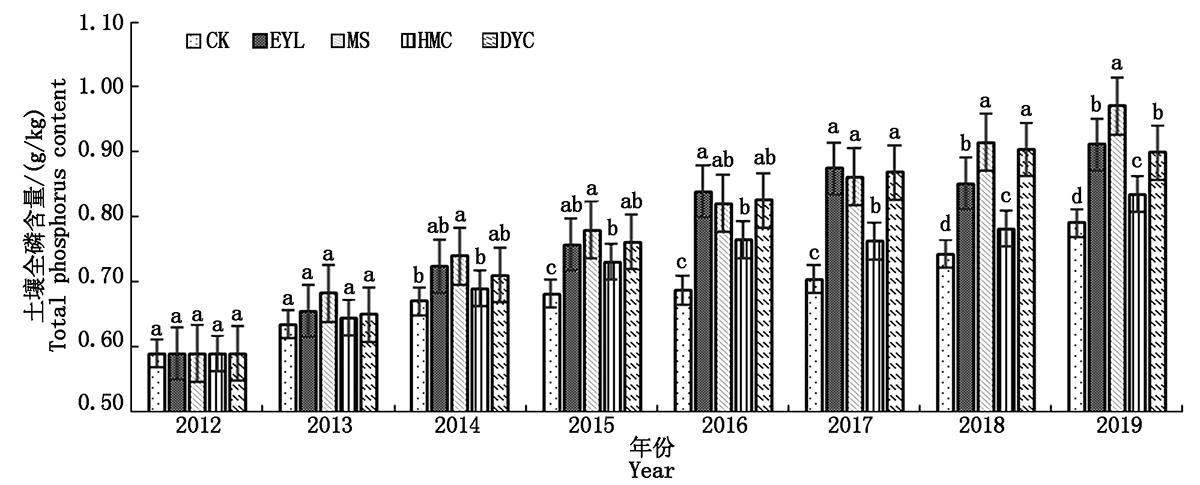
In order to promote the planting of winter green manure in North China,a long-term field experiment was adopted,and the winter idle field was used as a control.Four winter green manure treatments were set up,Orychophragmus violaceus,Viciavilosa Roth,Lolium perenne L.and Brassica campestris L..The effects of different winter green fertilizers on soil organic phosphorus and phosphorus content and corn phosphorus absorption were studied.The results showed that in the past 7 years of different winter green manure treatments,the change trend of soil total phosphorus and available phosphorus content was the same.Compared with other treatments,vetch increased the soil total phosphorus the most,with an average annual growth rate of 8.07%,winter rape increased soil availability compared with other treatments.The phosphorus content was the most,with an average annual growth rate of 48.34%.Different winter green manures and spring corn rotations had different effects on the organic phosphorus content of various forms in the soil.In 2019,the highest active organic phosphorus content was ryegrass treatment and winter rape treatment,both of which were 12.64 mg/kg;moderately active organic phosphorus content the highest was the winter rape treatment(41.64 mg/kg),the medium-stable organic phosphorus content was the highest in the ryegrass treatment(18.73 mg/kg),for the high-stability organic phosphorus,the winter rape treatment was significantly higher than other treatments.The treatments of February orchid,vetch and ryegrass were significantly lower than the treatments of winter fallow and winter rape;the rotation of winter rape and spring maize could promote the conversion of organic phosphorus to active and medium active organic phosphorus in the soil.After 7 years of winter green manure,the phosphorus content(0.39 mg/kg)of corn kernels treated with winter rape was the highest among the treatments.The order of correlation coefficients between the phosphorus content of corn grains and various forms of organic phosphorus was MLOP(0.952**)>LOP(0.816**)>MROP(0.122)>HROP(-0.064).
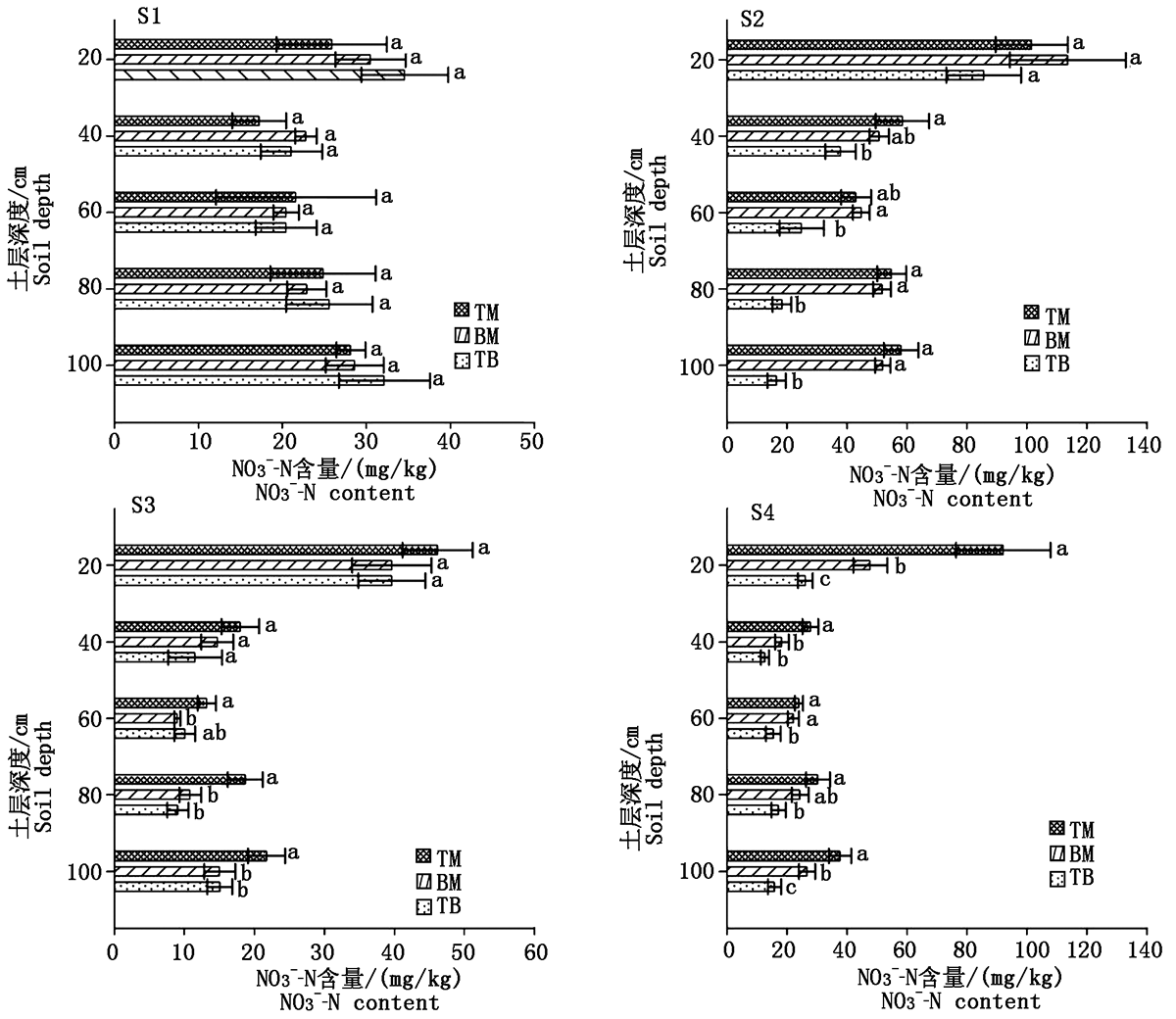
To investigate the effect and mechanism of different rotation systems on nitrate leaching,field experiments were carried out from 2018 to 2020 to compare the variation characteristics of nitrate leaching in greenhouse soil under three rotation systems,i.e.tomato-melon(TM),bean-melon(BM)and tomato-bean(TB),and to explore the controlling factors for differences in nitrate leaching.Results showed that the rotation with Vigna unguiculata significantly reduced the amount of nitrate leaching compared with the traditional TM rotation.The total nitrate leaching loss of TB was significantly decreased by 39.74% compared with TM,however,the nitrate leaching loss of BM were significantly decreased by 6.32%.Collectively,TB had the best environmental benefits among three rotations,which was the recommended rotation pattern.Spearman correlation analysis showed that the NO3-NO3--N leaching was most affected by 0—100 cm soil water storage,soil NO3--N accumulation and temperature,with a highly significant positive correlation.A positive correlation of NO3--N leaching with 0—100 cm organic carbon storage and soil total nitrogen accumulation,as well as a negative correlation with 0—60 cm soil pH value were also observed.Comparing with the traditional TM,the decreasing of nitrate leaching in recommended rotation pattern(TB)was mainly attributed to significant reduction of 0—100 cm soil water storage,soil NO3-N and total nitrogen accumulation,and significant increment of soil pH value to chang soil physical and chemical properties,together with alleviating the background nitrate leaching caused by organic nitrogen mineralization in sensitive leaching seasons.
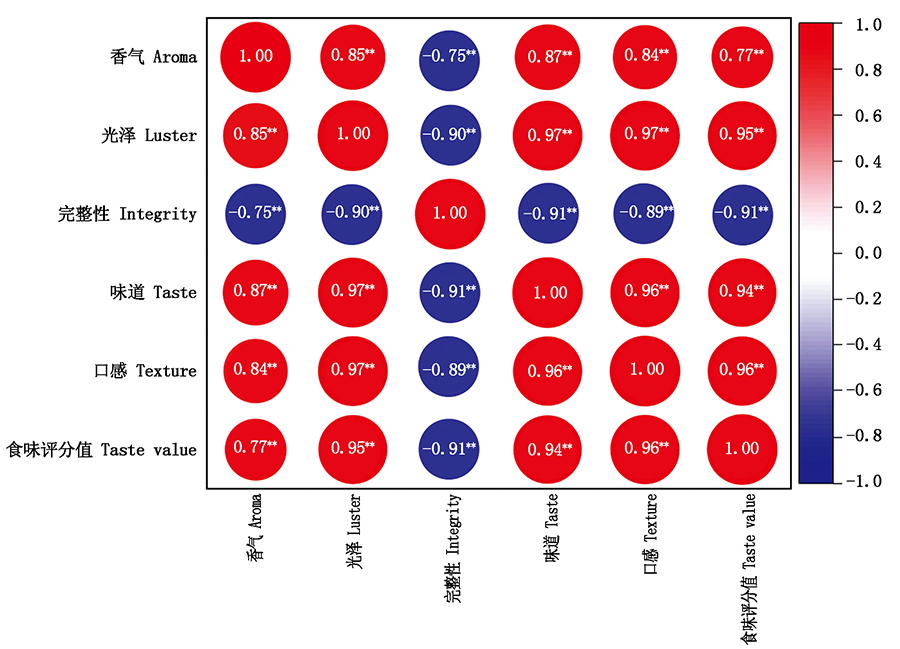
In order to explore the changing law of dry direct-seeding rice quality under different organic fertilizer treatments,this experiment was conducted from 2018 to 2020.Longjing 31 was used as the test material.Treatments includes:zero fertilizer(N0),conventional fertilization(NPK),Biochar+conventional fertilization(OF1),seaweed bio-organic fertilizer+conventional fertilization(OF2),Jishiwang bio-organic fertilizer+conventional fertilization(OF3),attapulgite organic fertilizer+conventional fertilization(OF4).Changes in the processing quality,appearance quality,nutritional quality,and cooking and eating quality of dry direct-seeding rice were investigated.Compared with NPK,the head rice percentage of OF1,OF2,OF3,OF4 increased by 2.64 ,1.78,1.06,2.53 percentage points,respectively.The chalkiness degree of OF2 was the highest(1.82%),which was 0.11 percentage points higher than that of NPK.However,the chalkiness degree of OF1,OF3 and OF4 were lower than that of NPK,with an average decrease of 0.26,0.41,0.51 percentage points,respectively.There was no significant difference in grain length,grain width and length width ratio among the treatments.From 2019 to 2020,compared with NPK,organic fertilizer treatment increased protein content,but decreased amylose content;the protein contents of OF1,OF2,OF3 and OF4 were 8.99%,9.13%,9.08% and 9.16% respectively,which were 0.54,0.68,0.63,0.71 percentage points higher than those of NPK;the amylose content of OF1,OF2,OF3 and OF4 decreased by 0.20,0.64,0.69,0.38 percentage points,respectively;compared with NPK,the application of organic fertilizer at the initial stage could improve the luster,taste,flavor and taste value of dry-direct seeding rice,but long-term application of organic fertilizer would lead to poor cooking and eating quality;from 2019 to 2020,the average taste value of NPK treatment was 72.70,compared with NPK,OF1,OF2,OF3 and OF4 reduced the aroma,luster,taste,flavor and taste value of dry direct-seeding rice by 5.53%,6.40%,3.71% and 3.23%,respectively,but organic fertilizer increased the integrity of rice.In conclusion,long-term application of organic fertilizer can improve the processing quality,appearance quality and nutritional quality of dry direct-seeding rice,but it is not conducive to the formation of cooking and eating quality and reduce amylose content.
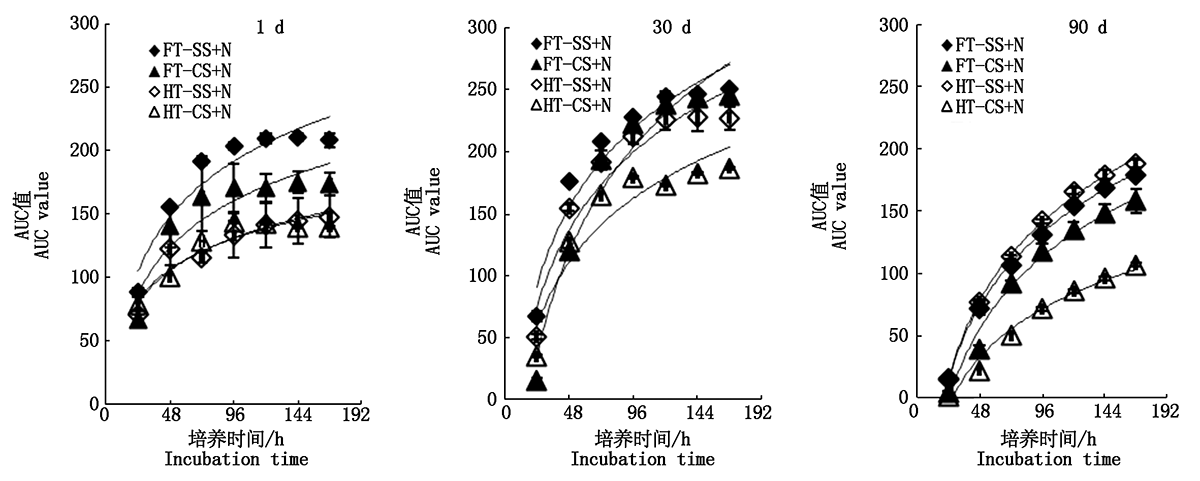
To study the decomposition characteristics of sorghum and maize residues and the functional diversity of microbial community in straw decomposition,under different residual types(sorghum stalk-leaves,maize stalk-leaves,sorghum root,maize root),soil type(cinnamon soil,yellow loam soil),nitrogen treatment(adjust C/N ratio,not adjusted),the culture experiment method was used to explore the decomposition characteristics of residues under different decomposition conditions,and to analyse the metabolic functional diversity of microbial community used the BIOLOG-ECO plates method in straw decomposition process.The results showed that the decomposition rate of sorghum and maize residues was faster in the early stage than the late stage.The residual dry matter degradation rate and CO2 release rate had the following pattern of decomposition rate was shown,sorghum stalk-leaves>maize stalk-leaves>sorghum root>maize root,under the same soil and nitrogen treatment.According to the second experiment, the dry matter degradation rates of sorghum stalk-leaves and maize stalk-leaves were 55.50% and 48.00%,respectively,however,the dry matter degradation rate of sorghum root and maize root were 31.25% and 16.75%,respectively at 60 d of incubation,under the condition of cinnamon soil+N treatment.The degradation rate of hemicellulose and cellulose in root were lower than that in stalk-leaves,under the same soil and N treatment conditions,the decomposition of hemicelluloses and cellulose was decreased by 23.70,18.80 percentage point,in sorghum root treatment than sorghum stalk-leaves treatment at 90 d of incubation,under the condition of cinnamon soil+N treatment.The microbial metabolic activity of straw was the highest at 30 d and lowest at 90 d during decomposition.The metabolizable ability of microbial community of straw decomposition to amines and phenolic acids was lower on the 1st day of decomposition,and the metabolizable ability to carbohydrates,amino acids and polymers was significantly decreased on the 90th day,compared with 30 d.Collectively,the decomposition of sorghum residues was easier than maize,and adjusting C/N ratio could accelerate the decomposition of residue to a certain extent.The microbial metabolic diversity was the highest at 30 d under the experimental conditions.
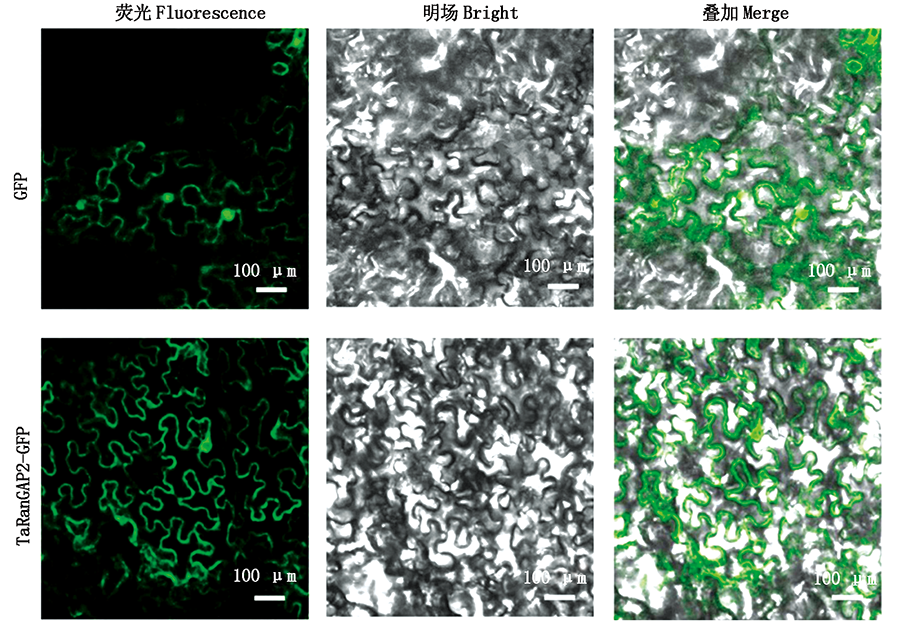
The aim to study the role of TaRanGAP2 in HR induction of wheat resistance to Puccinia triticina,so as to lay a foundation for elucidating the mechanism of Puccinia triticina resistance and breeding for disease resistance in wheat at the molecular level.The wheat near-isogenic line TcLr26 and its recurrent parent Tatcher(Tc)were used to form incompatabile(TcLr26×260)and compatabile combination(Tc×260)with Puccinia triticina race 260,respectively.Bioinformatics analysis showed that the total length of CDS of TaRanGAP2 was 1 665 bp,encoding 554 amino acids.Analysis of its conserved domains revealed that the protein encoded by TaRanGAP2 gene belonged to the Ran GTPase family;RT-qPCR was used to detect the expression of TaRanGAP2 in the two combinations.It was found that the expression of TaRanGAP2 was significantly up-regulated in the incompatible combination;the subcellular localization detection of TaRanGAP2 by tobacco transient transformation system showed that TaRanGAP2 was localized in the cytoplasm;Virus induced gene silencing(VIGS)technology were used to silence TaRanGAP2 gene in TcLr26.After inoculation with Puccinia triticina race 260,it was found that HR area of TaRanGAP2 gene-silencing plants was significantly increased,and the number of HMCs were also significantly increased compared with the unsilenced plants control plants.These results proved that TaRanGAP2 played a positive regulatory role in the induction of HR in wheat resistance to Puccinia triticina.
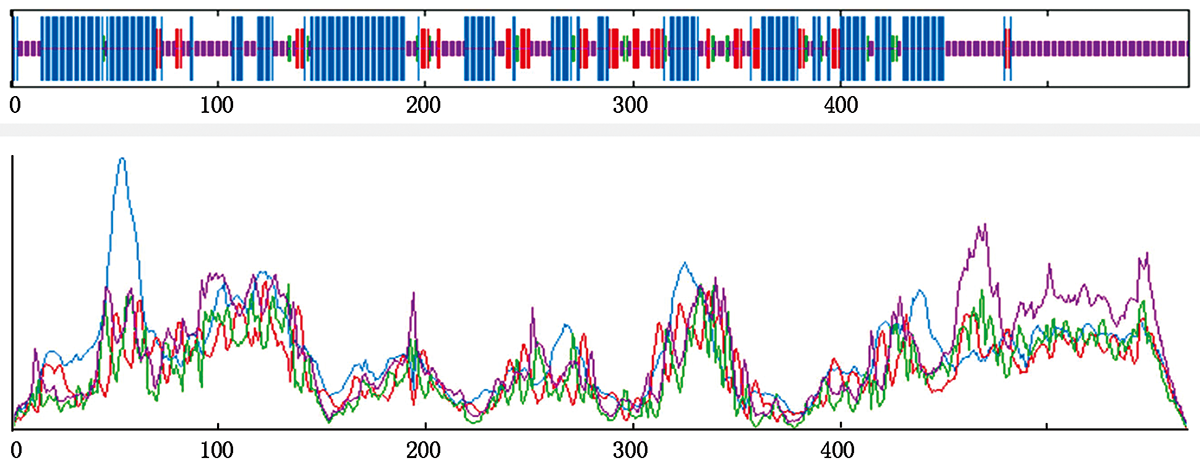
MLO gene,acting as susceptibility factor,play an important role in regulating responses of host plants against powdery mildew.To determine the gene function of bitter gourd McMLO1 gene,cloning,bioinformatics and expression analysis were carried out in the present study.The results suggested that the full-length of McMLO1 was 4 019 bp,among which the CDS sequence was 1 707 bp,encoding 568 amino acids.ProtParam prediction indicated McMLO1,whose molecular weight and theoretical isoelectric point was 65.40 ku and 9.36,respectively,was an unstable hydrophilic protein which located in cell membrane.McMLO1 protein,harboring a conserved MLO domain of 477 amino acids,was consisted of random coil and alpha helix on secondary structure.Multiple sequence alignment and phylogenetic analysis revealed high homology between McMLO1 and cucumber MLO protein.qRT-PCR analysis suggested that the expression level of McMLO1 in leaf was much higher than that in other tissues.Moreover,the expression level of McMLO1 was significantly up-regulated and reached peak at 6 hours after inoculation with powdery mildew pathogen,indicating McMLO1 participate in the process in response to powdery mildew in bitter gourd.
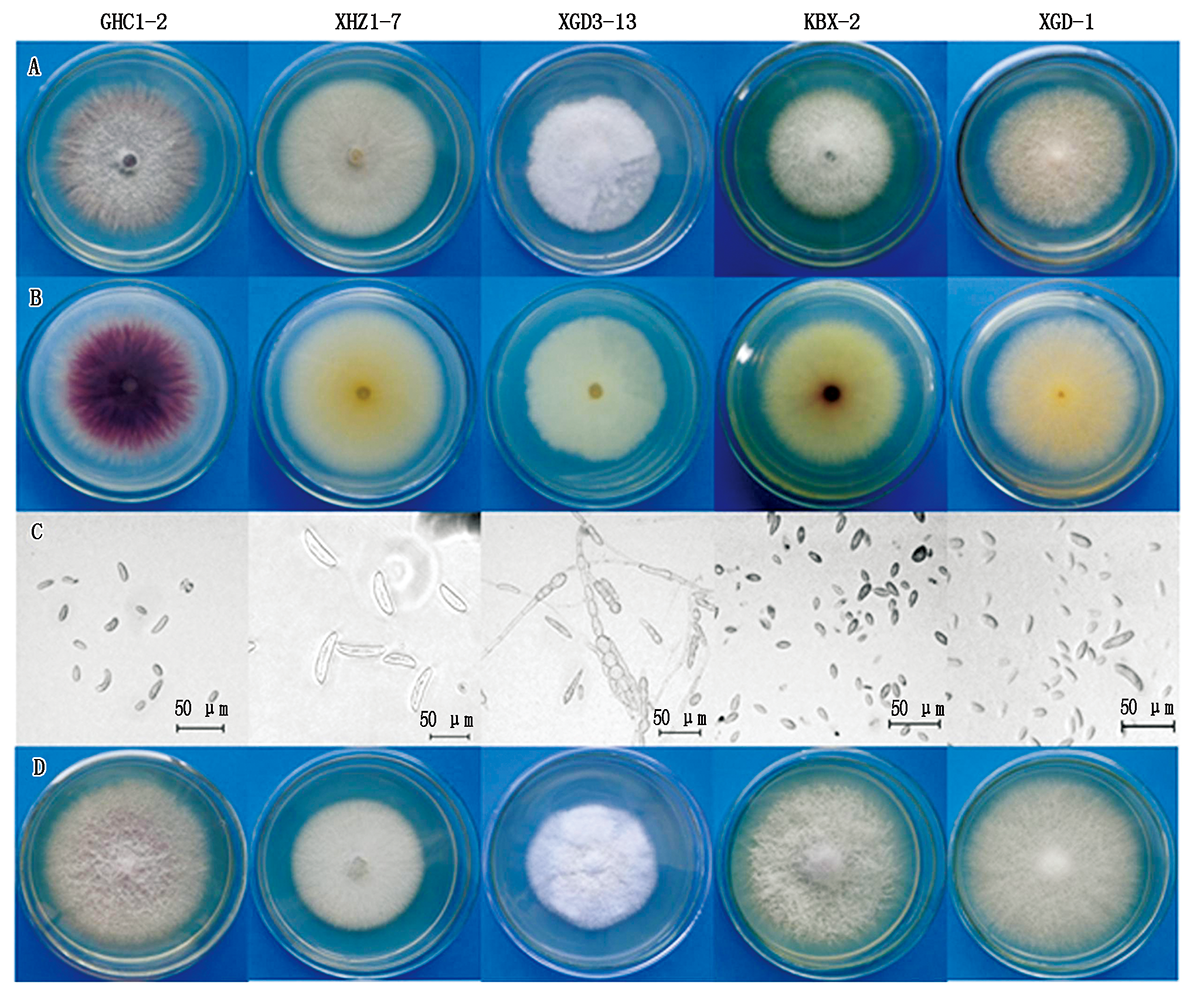
In order to obtain the biocontrol strains of sunflower broomrape,this study took the occurrence of necrotic spot as samples of sunflower broomrape,and used the Koch's rule to separate and identify 29 samples of necrotic spot collected from 11 different locations in Inner Mongolia,Hebei and Xinjiang.The results showed that Fusariun oxysporum and F.solani were isolated from the disease samples collected from the 183 Regiment and 188 Regiment of Beitun Agricultural Tenth Division,Xinjiang and Xihaizi Village,Wuchuan County,Hohhot.F.oxysporum and F.equiseti were isolated from disease samples from Xixiaozhao Town,Urad Front Banner,Kuiboyuan,Wuyuan County and Yongli Village,Linhe District in Bayannur City.F.oxysporum,F.solani,F.equiseti and F.proliferatum were identified from the disease samples in the Gonghecheng Village and Xigedan Village,Siziwang Banner and a plot next to Provincial Highway 105 in Huade County.Only F.oxysporum was isolated from the samples collected in Dongsheng District,Ordos.In Kangbao County,Zhangjiakou City,Hebei Province,only F.verticillioides was isolated from samples collected.The above-mentioned isolated strains could cause disease symptoms after being re-inoculated on Helianthus angustifolia.The result indicated that the fungus of the genus Fusarium was the pathogenic fungi that caused blight in sunflower broomrape.However,F. oxysporum was the predominant pathogen of sunflower broomrape necrotic spot.The results of pathogenic differentiation studies showed that all the isolated strains were able to infect sunflower and broomrape,however,there were certain differences in their pathogenicity,which mainly exhibited stronger pathogenicity on isolated hosts than that of other host.Based on the results of pathogenicity differentiation,three strains of F.oxysporum GHC 1-2,XXZ-9 and HD 1-1 with biocontrol potential on broomrape were selected to control the parasitism of broomrape on sunflower.
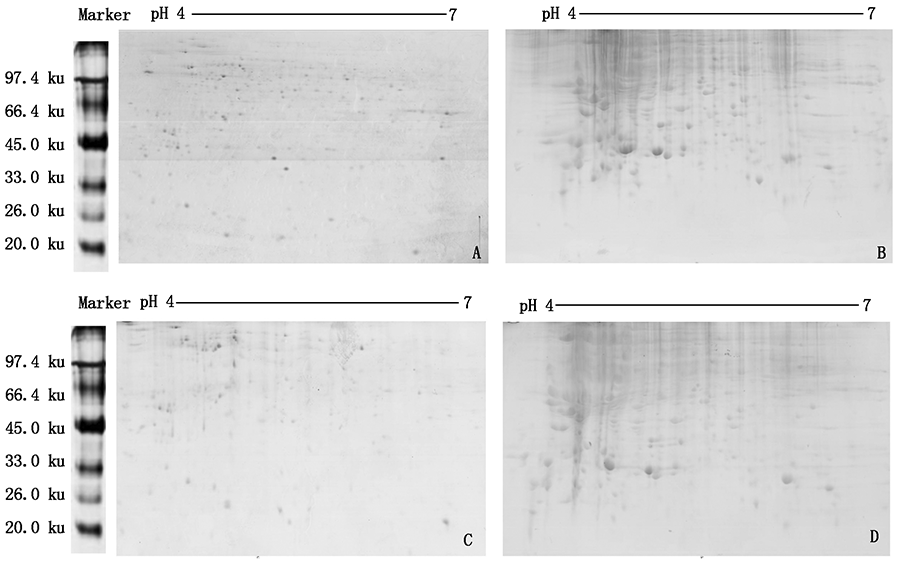
In order to establish a proteomic 2-DE system for the defense response of Cabernet sauvignon winter buds under Apolygus lucorum feeding stress,Cabernet sauvignon grape winter buds were used as materials to compare the effects of different protein extraction methods,sample loading and gel staining techniques on the 2-DE chromatogram.It provided reference for subsequent studies on screening and analyzing differential proteins.Winter buds of Cabernet sauvignon grape were selected,and feeding stress and blank control experiments were conducted,respectively.After 24 h,the samples were taken back with liquid nitrogen and frozen at -80 ℃.Crude protein powder was extracted by TCA-acetone and TCA-acetone+phenol extraction.Protein concentration was determined by CBB G-250 staining.Sample loading and gel staining techniques were determined.Gel images were obtained by scanning for comparative analysis.Crude protein for 2-DE could be obtained by different extraction methods.The amount of crude protein obtained by TCA-acetone method was significantly higher than that obtained by TCA-acetone+phenol extraction method.The concentrations of total protein extracted by the two methods were different,but both of them met the requirements of two-dimensional gel electrophoresis.TCA-acetone method was easier to operate and less protein loss.The influence of different loading amount on the effect of 2-DE was significant.Compared with 800 μg protein,400 μg protein showed clear protein spots and was easy to separate.The results of CBB staining showed that R-350 was more suitable for total protein two-dimensional gel electrophoresis of grape winter bud.Using TCA-acetone method,400 μg protein sample,17 cm pH value 4—7 IPG dry adhesive strip,G-350 thermal staining 2-DE system to obtain the total protein gel map of Cabernet sauvignon winter buds met the requirements of further test.
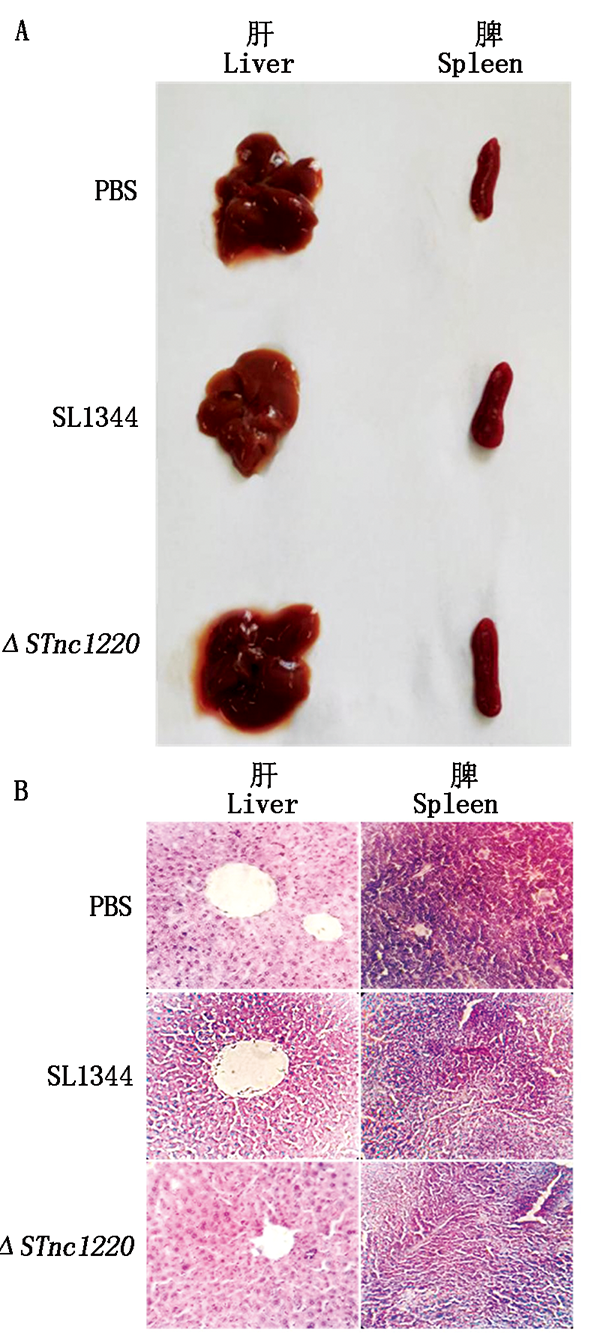
In order to explore the molecular characteristics,biological functions and effects of sRNA STnc1220 of Salmonella typhimurium on bacterial virulence,the sRNA STnc1220 gene of Salmonella typhimurium was amplified by PCR,and its molecular characteristics were analyzed.Use the TargetRNA2 website to predicted the target genes that might be regulated by the STnc1220 gene;the λ-Red homologous recombination technology was used to construct STM(Salmonella)-The gene deletion mutant strain,and compared with the parent SL1344,its growth characteristics,biochemical characteristics,genetic stability,biofilm(BF)formation ability,and its effects in different stress environments(pH,H2O2,high salt and ethanol stress)adaptability,through adhesion and infection of macrophages and mouse infection tests to study pathogenicity.The results showed that the full length of STnc1220 gene was 73 bp,and its upstream 5'-UTR had-10 and-35 promoter binding sites.It was predicted that its potential target gene was barA gene;The test successfully constructed the deletion strain STM-ΔSTnc1220,and it had good performance genetic stability;compared with the SL-1344 parent strain,the growth rate,biochemical characteristics and biofilm formation ability of the missing strain did not change;the growth rate was not significantly different under the conditions of pH=4.8,pH=9 and H2O2.However, in the 4% NaCl environment, the growth rate of the deletion strain was significantly higher than that of the parent strain after 7 h, and the growth rate of the deletion strain in the logarithmic phase was significantly increased in the 3.8% ethanol environment(P<0.05);It significantly reduces the ability of adhesion and invasion in macrophages(P<0.01), and the LD50 of mice was significantly elevated,the pathological damage to the liver and spleen of mice was weakened.It showed that the STnc1220 gene played an important role in environmental adaptation and virulence regulation.
To clone the full gene sequence of Mythimna separate serine protease,and express it in vitro,Mythimna separata was used as the research object,the partial gene of serine protease was amplified by RT-PCR,and further the whole gene was obtained by RACE technology.The obtained gene sequence and deduced protein sequence of serine protease were analyzed using Blast software and other softwares.The gene of serine protease was expressed using the Escherichia coli expression system.The results showed that the full serine protease gene from Mythimna separate was successfully cloned and named as MsPG,with the sequence length of 2 010 bp,and the open reading frame of 1 593 bp,encoding 530 amino acids.Its protein had the molecular weight of 67.6 ku,and the isoelectrical point of 7.63,and owned a clip-type domain containing 6 cysteine,which was speculated as a clip-type serine protease.The amino acid sequence of MsPG had identities of 70.00%—91.92% to those of other Lepidoptera insects,and showed the highest identity(91.92%)to that of Heliothis virescens(GenBank No.PCG78401.1).The recombinant plasmid pET30a(+)-MsPG was successfully constructed and expressed by 0.1 mmol/L IPTG induction in E.coli prokaryotic expression system,with the highest expression at 25 ℃.In conclusion,MsPG has conserved functional sites in the serine protease family and can be prokaryotically expressed in vitro.

The aim of this study was to investigate the effects of low protein diet on meat quality,amino acid and fatty acid composition of skeletal muscle and blood,and mRNA expression of myogenic regulatary factor genes of black goat.A total of 24 female Xiangdong black goats were randomly divided into control group(CON,10.77% CP,n=12)and low protein group(LP,5.52% CP,n=12).The pre and post-trial lasted for 25 d and 45 d,respectively.The results were as follows:Low protein diet increased L* 24 h of meat color,but had no difference on pH value and water loss rate.The percentage of type Ⅰ muscle fiber and type Ⅱ muscle fiber had no difference between two groups,only the average fiber cross sectional areas of semitendinosus had a tendency to reduce.Valine,leucine,lysine,phenylalanine,glutamate and total essential amino acids of vastus lateralis significantly reduced,isoleucine,methionine,aspartate,arginine and total amino acid tended to decrease.Glutamate of semitendinosus in the LP group significantly reduced,threonine and serine tended to decrease.Compared with the control group,C20:2 and C22:1n9 of semitendinosus in the LP group significantly decreased.Total PUFA,C18:1n9c,and C20:4n6 of vastus lateralis in the LP group significantly decreased;C18:1n9c of longissimus dorsi in the LP group significantly decreased.Compared with the control group,the mRNA expression of MYOD in semitendinosus were significantly down-regulated in the CON group.In conclusion,low protein diet didn't affect type conversion and cross sectional area of muscle fiber.Dietary protein deficiency reduced the deposition of amino acid in muscle,while reduced the deposition of polyunsaturated fatty acids in muscle,which affected on muscle flavor of goat.

The aim of this study was to obtain the CDS of CIDEB and CIDEC gene,detect the expression of CIDEB and CIDEC in different tissues of goats,predict their interaction proteins.It would provide basial data for revealing the regulatory role of CIDEs in lipid metabolism.The CDS of CIDEB and CIDEC gene were cloned by RT-PCR,and the biological characteristics and interaction proteins of CIDE proteins were analyzed by online tools.Real-time quantitative PCR(RT-qPCR)was used to detect the expression of CIDEs and the genes,encoding the interaction proteins,in different tissues of goats,following the correlation analyses by a SPSS software.The CDS of CIDEB was 660 bp,encoding 219 amino acids,while the CDS of CIDEC was 714 bp,encoding 237 amino acids.Phylogenetic tree analysis showed that goat CIDEA,CIDEB and CIDEC were closely related to corresponding genes from sheep.RT-qPCR detection showed that CIDEA,CIDEB and CIDEC were highly expressed in the rumen,liver and small intestine of goat,respectively.The expression of CIDEA was significantly correlated with the expression of DFFB (P<0.01),ELOVL3 (P<0.01),DIO2 (P<0.05),TMEM26 (P<0.05),PRDM16 (P<0.05)and PLIN1 (P<0.05)in rumen.The expression of CIDEB was significantly correlated with that of DFFA and SDR39U1 in liver(P<0.05).The expression of CIDEC was obviously correlated with that of PLIN2,GPLD1 and ADIPOQ in small intestine(P<0.05).Through the analysis,we found genes related to the expression level of CIDEs,which could provide a theoretical basis for further revealing the role of CIDE gene in lipid metabolism and its regulatory network.Moreover,the proteins of CIDEs were all lipid-droplet related proteins,and could also provide a reference for further studying the precise mechanism of lipid droplet formation.
This aim of study was to explore the role of ORFV118 in regulating cell cycle,apoptosis and the immune-related cytokines(IL-1β,IL-6,IFN-γ and TNF-α)in goat testicular sertoli cells.We cloned the whole sequence of ORFV118 and constructed an expression vector of pEGFP-ORFV118.The expression of ORFV118 protein was then identified by using Western Blot method after the transfection of goat testicular sertoli cells with pEGFP-ORFV118.Following which,a flow cytometer equipment was used to measure the effect of ORFV118 expression on cell cycle and apoptosis of goat testicular sertoli cells,and also on regulating the expression of genes related to cell cycle,including CDK2 and P21,and genes relative to apottosis,including Bax,Bcl-2,Caspase3,Caspase7,P53 and BCL2L11.The expression of immune-related cytokines,including IL-1β,IL-6,IFN-γ and TNF-α,was detected by ELISA method and RT-qPCR after ORFV118 expression in goat testicular sertoli cells.The results showed that a total length of 309 bp ORFV118 gene sequence was cloned successfully.The cell's DNA synthesis phase(S phase)was inhibited and the late stage DNA synthesis(G2/M phase)was promoted by the expression of ORFV118,and the expression of CDK2 mRNA was decreased.The cell apoptosis blocked after ORFV118 expression,and the expression of Caspase3,Caspase7 and SOCS2 mRNA decreased.The immune-related cytokines,including IL-1β and IFN-γ,was inhibited in goat testis cells,while the expression of IL-6 and TNF-α were promoted.
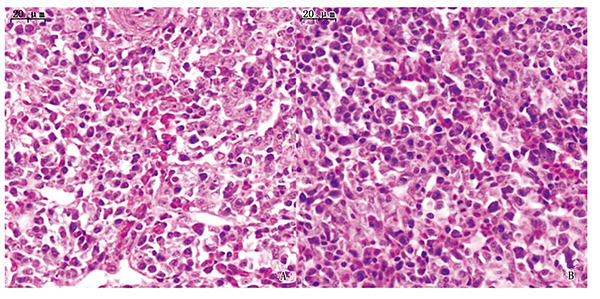
In order to study the effect of the effector protein Hcp2b on the transcriptomics of the spleen during the infection of Avian pathogenic E.coli(APEC)in chicks.The hcp2b-deletion strain Δhcp2b and the reverted strain Chcp2b(with AE17 strain as a positive control)constructed and preserved in the Anhui Province Key Laboratory of Veterinary Pathobiology and Disease Control were used to inject 7-day-old chicks intramuscularly.After 24 h,the spleen of depressed chicks was collected to detect the tissue load and make pathological sections.The deletion strain Δhcp2b and the wild strain AE17 were selected to infect spleen tissue for transcriptomics sequencing.Also,Real-time PCR was used to identify the sequencing results.Moreover,GO analysis and KEGG pathway enrichment analysis were performed on the differentially expressed genes.The results illustrated that the wild strain AE17,the deleted strain Δhcp2b and the reverted strain Chcp2b had no significant difference in the tissue load of the spleen.Observation of tissue sections that the spleens of wild strain AE17 and deletion strain Δhcp2b both suggested enlarged interstitial spaces and hyperemia.Transcriptomics analysis revealed that the deletion strain Δhcp2b induced the differential expression of spleen gene mRNA in chicks.515 differentially expressed genes were screened(185 genes were up-regulated and 330 genes were down-regulated).Real-time PCR confirmed that the expression of IL22,TNFRSF6B,and TNFRSF8 genes in the spleen was down-regulated after the deletion strain Δhcp2b infected chicks,which was consistent with the trend of the sequencing results.At 24 h,GO analysis found that the chick spleen differential genes were significantly enriched in items such as membrane,membrane composition,and extracellular space.KEGG analysis found that differential genes in the spleen were significantly enriched in the pathway of endoplasmic reticulum protein processing.The hcp2b gene had no effect on APEC colonization of spleen,the hcp2b participates in the pathogenic process through the signal pathway that affects the endoplasmic reticulum protein processing of the spleen in the body's immune organs.

Bimonthly, Started in 1962
CN 13-1101/S
ISSN 1000-7091
CODEN: HHUOA6
Responsible Institution: Hebei Academy of Agriculture and Forestry Sciences
Sponsored by: the Academy of Agricultural Sciences and Agricultural Association of Hebei, Beijing, Tianjin, Shanxi, Henan and Inner Mongolia.
Editor-in-chief: Qiang Zhang
Edited and Published by: Editorial Department of Acta Agriculturae Boreali-Sinica

《Acta Agriculturae Boreali-Sinica》Official Website

Wechat Official Account

Copyright © 2022 《Acta Agriculturae Boreali-Sinica》