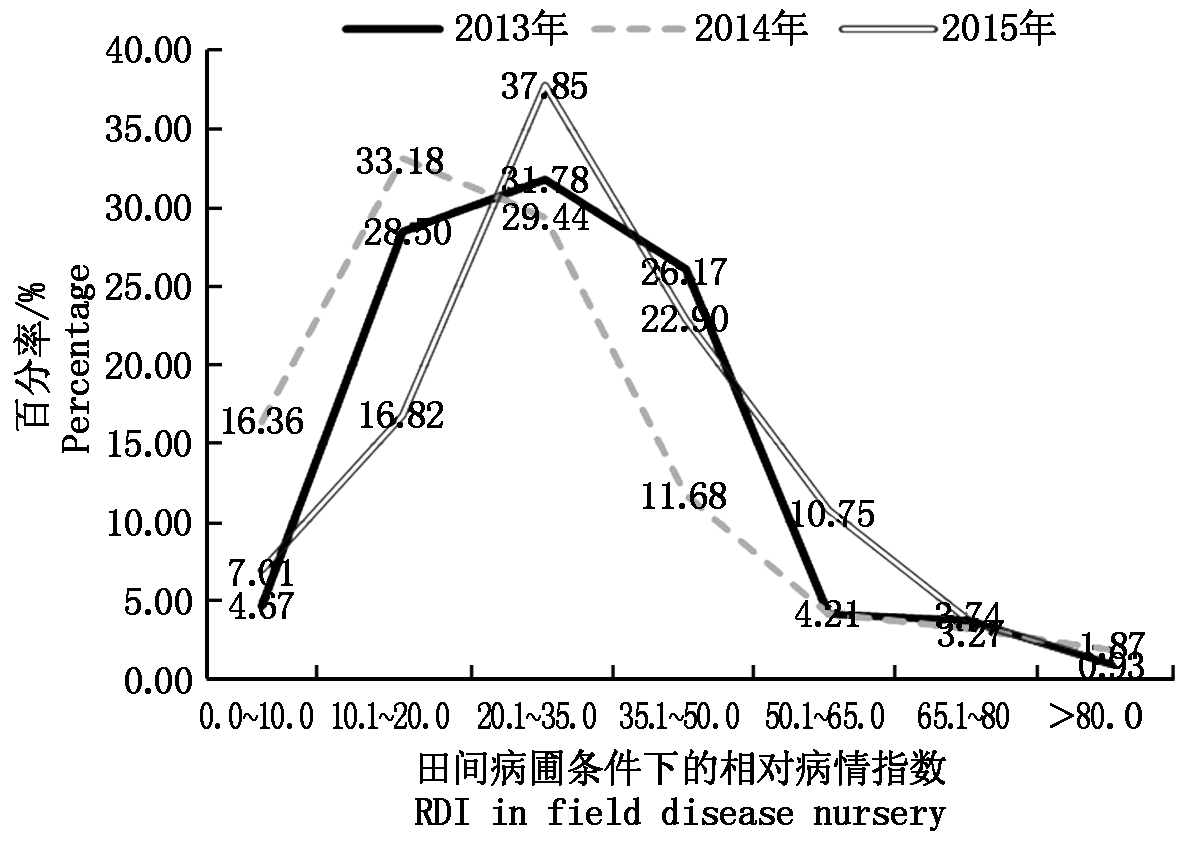
图1 田间病圃鉴定条件下供试材料病指变化频率分布
Fig.1 Histogram of relative disease index of tested lines identified in field disease nursery
陆地棉(Gossypium hirsutum L.)是世界上最重要的天然纤维作物,也是我国重要的经济作物之一[1]。黄萎病是一种严重的土传病害,寄主范围广泛[2]。目前,国内缺乏高抗黄萎病的陆地棉品种,且没有有效治愈黄萎病的杀菌剂,使得黄萎病成为危害棉花的最大病害之一[3-4]。2009-2010年,超过我国棉花种植面积的50%受到黄萎病的影响(美国国家棉花委员会-疾病数据库)[5]。培育和推广抗病品种是防治棉花黄萎病的有效方法[3]。
由于当前陆地棉品种遗传背景狭窄,且抗病性和产量、纤维品质之间存在一定程度的遗传负相关性[6],实现三者的同步改良是一项艰巨的考验。分子数量遗传学发展为实现遗传改良提供了有效的工具,通过挖掘抗病性相关的QTLs(Quantitative trait loci)应用于分子辅助育种可加速选育抗病品种的进程。
为了挖掘抗病性相关的QTLs,不同研究者利用2个不同黄萎病抗性的棉花材料构建分离群体,通过连锁作图定位到众多与抗黄萎病性状相关的QTLs[7-13]。同时,随着遗传学的发展,基于连锁不平衡的关联分析在植物基因组学研究领域取得了成功的应用[14]。与连锁作图相比,全基因组关联分析(Genome-wide Association Study,GWAS)具有精确度高、不需专门构建群体、同时分析同一位点的多个等位基因等优点[15-17]。
近年来,关联分析方法已开始应用于棉花黄萎病抗性的研究。郭志军等[18]采用 74 个 Simple sequence repeat(SSR)标记对 172 份陆地棉栽培种的基因组变异进行扫描,通过关联分析发现 12个与黄萎病抗性显著相关(P<0.05)的位点;李黎贝[19]利用140 个SSR标记在 186 份陆地棉材料中共检测出 355 个等位变异,挖掘与棉花黄萎病抗性状相关联的位点22个(P<0.01)。Zhao等[20]搜集了世界各地的158份陆地棉资源,在全基因组内均匀筛选212个多态性较好的SSR标记,在成株期和苗期对陆地棉的黄萎病抗性进行了鉴定,利用关联分析挖掘了42个与黄萎病抗性关联的位点,其中有30个为首次挖掘,此外,本研究在16号染色体上发现了与黄萎病抗性相关的QTL簇。目前,通过关联分析挖掘陆地棉黄萎病抗性相关位点取得了一定的进展,但是与棉花其他性状比如纤维品质、产量相比[1,6,21-24],相关研究尚不充足。
本研究通过分析214份陆地棉资源的遗传多样性,利用黄萎病病圃对其多年发病情况进行调查,利用均匀分布于棉花基因组上的237个SSR标记进行全基因组扫描,发掘与棉花黄萎病抗性显著关联的标记位点,旨在为棉花抗黄萎病性状的分子检测及标记辅助选择育种提供参考信息。
214份陆地棉资源均由河北省农林科学院棉花研究所分子育种室提供,包括我国陆地棉栽培种(107个)、搜集于国内的资源(55份)、国外的资源(7份)、本实验室自选的育种材料(45个)。这些材料来源广泛,表型性状比如株高、纤维品质、熟性、衣分、产量、叶色、株形等方面差异显著。2013-2015年将上述材料种植于河北省农林科学院棉花研究所小安舍试验站田间病圃(河北石家庄),该田间病圃是多年植棉又经过人工改造的黄萎病病田,经多年田间观察统计,具有与黄萎病病圃相当的致病效果。试验采用3行区,行长7.5 m,行距0.8 m。定苗时每行留27株左右。采用局部随机排列,设3个重复。组内每隔10个品种设感病对照品种(冀棉11)一次。
黄萎病鉴定采用田间成株期鉴定,参考国家棉花品种区试中抗黄萎病的鉴定方法[25]。根据黄萎病发病高峰期分别于每年8月底至9月初进行田间鉴定,每重复取中间行20株调查黄萎病发病情况,感病对照为冀棉11,采用3个重复相对病情指数(Relative disease index,RDI)平均值表示各个材料的抗病性强弱[25]。
CTAB法提取基因组DNA[26],采用1%琼脂糖检测DNA完整性和杂质,采用分光光度计Nanodrop 2000检测,稀释至终浓度25~40 ng/μL。
本试验从6 000余对SSR引物中选取237个扩增效果稳定、多态性好且分布在棉花26条染色体上的SSR标记用于群体扩增,引物在染色体上的位置参考Liang等[27]构建的连锁图谱。所有引物信息均可从CottonGen(http://www.com.cottongen.org/)和CMD(http://www.cottonmarker.org/)查取。PCR(Polymerase chain reaction)反应体系10 μL,模板DNA 1.2 μL,天根2×Taq RCR Master Mix 5 μL,上下游引物各0.5 μL,ddH2O 3 μL。PCR反应程序为:95 ℃预变性4 min;94 ℃变性45 s,55 ℃退火45 s,72 ℃延伸1 min,30个循环;72 ℃ 10 min,8%的垂直板非变性聚丙烯酰胺凝胶电泳分离,参考张军等[28]方法银染。
人工读带,同一位置上清晰且重复性好的记为“1”,无带记为“0”,带弱或模糊不清的记为缺省值;利用Powermarker 3.25[29]计算引物的多态性位点、多态性信息含量和多样性指数,并利用其Phelogeny功能,计算Nei氏遗传距离,构建群体材料的Neighbor-Joining(NJ)聚类图;利用Structure 2.2 软件[30]估测该自然群体遗传结构,计算获得Q值。K值为1~10,将MCMC(Markov chain monte carlo)开始时的不作数迭代(Length of burn-in period)设为10 000次,采用Evanno等[31]提出的ΔK值方法确定合适K值;利用Tassel 2.1软件的一般线性模型(General linear model,GLM)进行关联分析,并计算各标记对表型变异的解释率[32]。
对214份陆地棉资源进行抗病性鉴定表明,陆地棉在黄萎病抗性上表现出广泛的变异。在田间病圃鉴定条件下,2013年的RDI变化为5.6~91.6,平均为30.6,2014年的RDI变化为1.5~98.3,平均为26.0,2015年的RDI变化为0~90.0,平均为33.3(图1)。方差分析表明:相同年份3个重复内的相对病情指数(RDI)不存在显著差异(P>0.5),而不同材料间RDI存在极显著差异(P<0.01)。不同年份之间进行比较发现,2014年黄萎病整体发病情况与其他2年相比较轻,年份效应显著。3年均表现抗病(RDI<20)的材料有18份,其中,本实验室自选的育种材料6份(抗8、冀棉18选系、冀选系258、冀选系120、Z56-1系、JMX-20),栽培品种11份(冀棉958、中棉所69、冀151、冀122、新陆中44、德利农5号、仁和39号、中棉所40、冀优768、冀棉616、冀228),资源材料1份(马里4号)。
利用PowerMarker v3.25计算软件进行遗传多样性分析,237个SSR引物共检测到280个多态性位点,涉及695个等位变异,变异范围 2~6 个,平均等位变异数为2.479 0个(表1)。JESPR153、BNL169和BNL3442检测到的等位变异数最多为6个,多态性信息含量(PIC)变幅为0.004 9(NAU3816)~0.610 8(JESPR153),平均0.213 3;基因多样性指数变异为0.004 9(NAU3816)~0.665 5(JESPR153),平均为0.255 8。SSR引物在所选棉花品种中检测的等位变异数目和基因多样性的跨度较大,但平均值较低,说明所选择的棉花材料在基因组水平上变异比较丰富,但整体上所选陆地棉的遗传基础比较狭窄。
图1 田间病圃鉴定条件下供试材料病指变化频率分布
Fig.1 Histogram of relative disease index of tested lines identified in field disease nursery
表1 SSR 标记多态性信息统计
Tab.1 Statistics of SSR marker polymorphism information
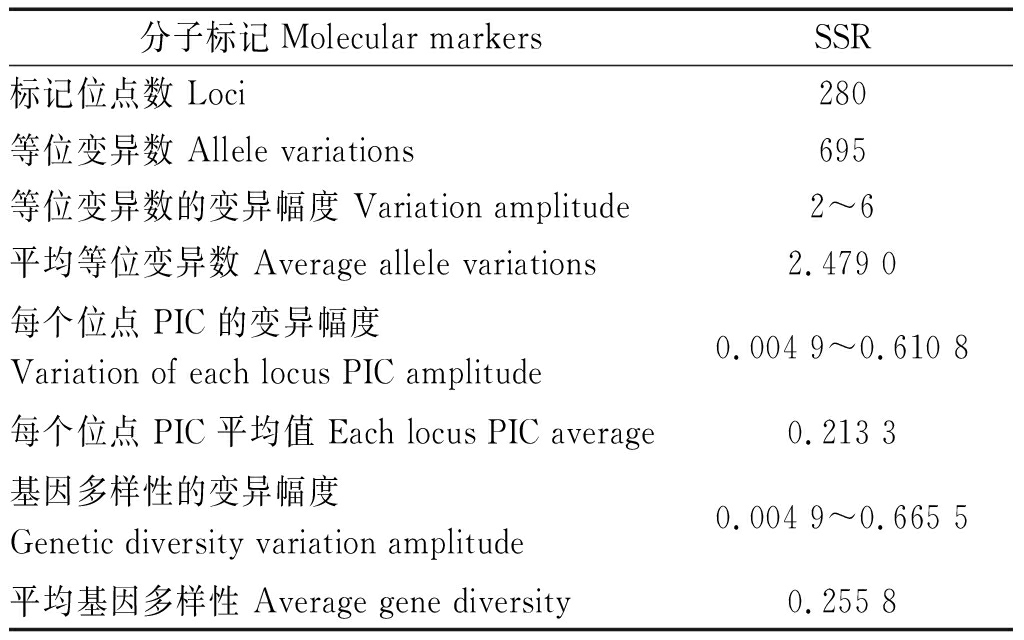
分子标记Molecular markersSSR标记位点数 Loci280等位变异数 Allele variations695等位变异数的变异幅度 Variation amplitude2~6平均等位变异数 Average allele variations2.479 0每个位点 PIC 的变异幅度 Variation of each locus PIC amplitude0.004 9~0.610 8每个位点 PIC 平均值 Each locus PIC average0.213 3基因多样性的变异幅度Genetic diversity variation amplitude0.004 9~0.665 5平均基因多样性 Average gene diversity0.255 8
利用PowerMarker v3.25软件中的Phelogeny功能,根据280个具有多态性位点分子标记数据,计算Nei氏遗传距离;并基于该遗传距离,构建群体材料的Neighbor-Joining(NJ)聚类图。采用Nei氏距离对该自然群体进行聚类分析(图2),可以分为3个亚群,分别包含8,79,127份材料。此分类与材料类型无对应关系。8份材料中有5份资源材料、3个品种,其中,7份材料在群体结构分析中划分为第1亚群;79份材料中有33份育种中间材料、31个品种和15份资源材料,在群体结构划分中均被划分为第1亚群;127份材料中有73个栽培品种、42份资源材料和12份育种中间材料,在群体结构划分中有70份划分为第1亚群,57份材料划分为第2亚群。聚类结果与材料的地域来源无显著对应关系,说明不同育种单位之种质流动性较大,打破了种质的地域界限。
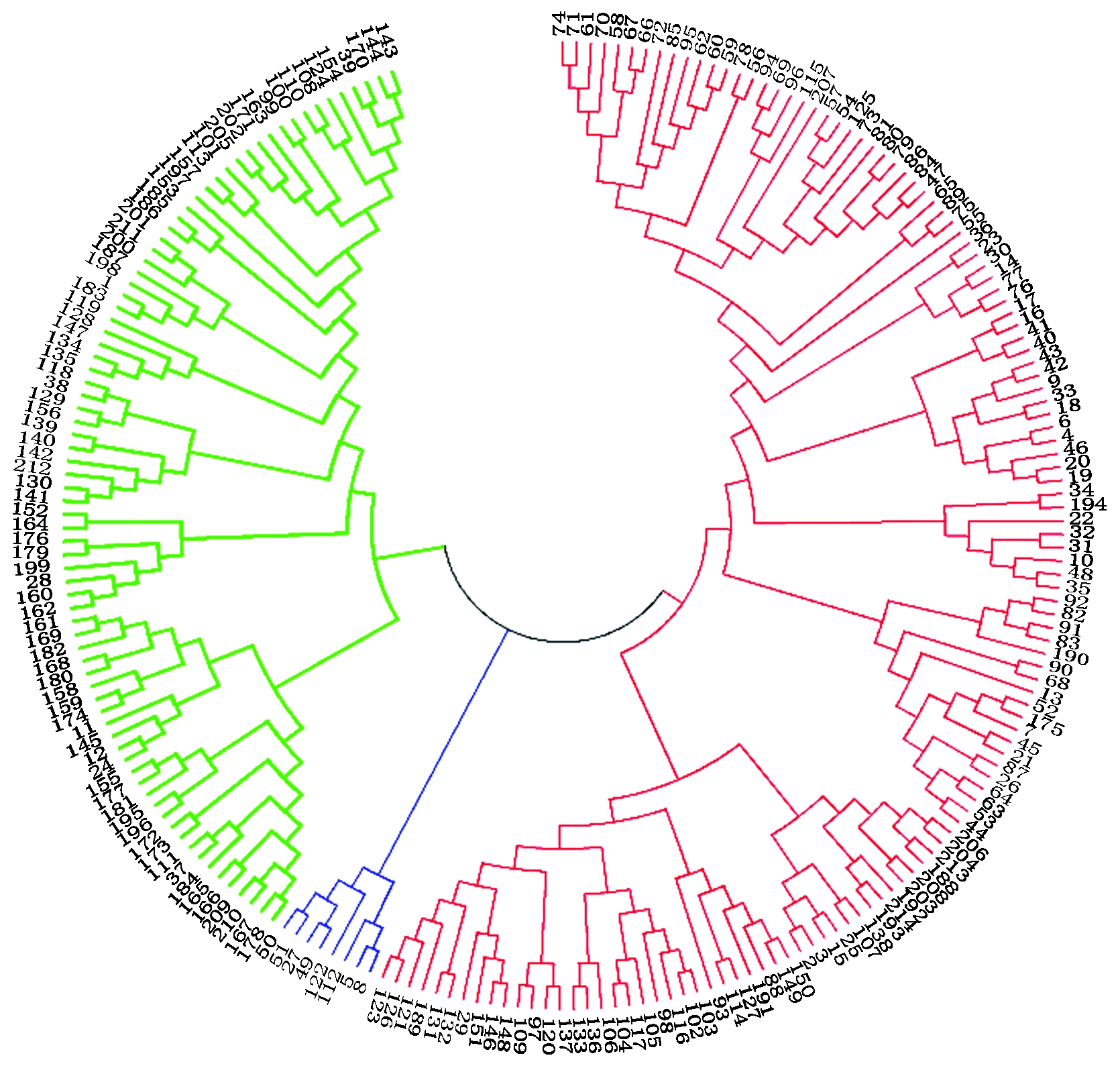
图2 214份陆地棉资源基于SSR标记的聚类
Fig.2 Clustering map of 214 upland cotton based on SSR markers
利用 Structure 2.2软件中的混合模型聚类分析法对214份陆地棉的群体结构进行划分,在K=1~10时,随着K值的增大,模型的后验概率(Lnp Posterior probability,Ln P(D))也持续增大,因而,无法依据最大似然值的原则确定合适的K值。参照 Evanno 等[31]的方法计算ΔK值,ΔK在K=2时出现顶点,推测该自然群体分为2个亚群(图3),2个亚群分别包含156,58份材料。此结果与利用NJ聚类方法分析结果略有差别,但共同表明供试群体结构简单,来源较为单一,在一定程度上反映了陆地棉品种遗传基础狭窄的问题。
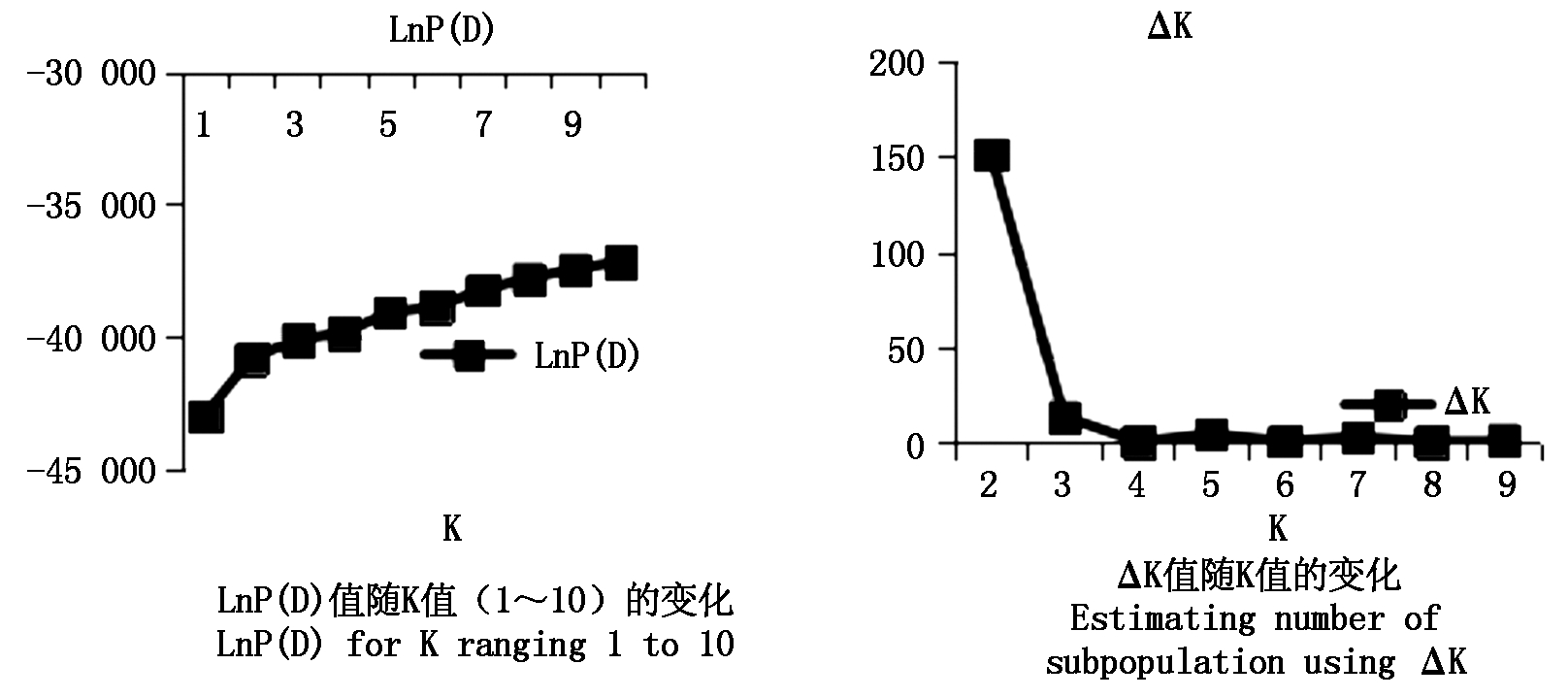
图3 214份陆地棉材料的群体结构
Fig.3 The population structure of 214 upland cotton germplasm
利用Tassel 2.1进行连锁不平衡分布(LD)分析,共有204个标记覆盖了整个棉花基因组2 918.7 cM,标记间的平均距离为14.38 cM,平均每条染色体上有7.85个位点,另有33个标记尚未定位到染色体上。237个标记在供试群体中检测到了280个多态性位点,这些位点间的连锁不平衡分布如图4所示。在280个位点的39 060种位点组合中,连锁不平衡位点广泛存在于共线性组合和非共线性组合中,概率统计极显著支持(P<0.01)的成对位点占总组合数的3.73%(图4),所选陆地棉基因组内的连锁不平衡水平较低。
通过关联分析,在不同年份中发掘与黄萎病抗性显著关联(P<0.01)的SSR位点27个(表2),其中有2个位点(BNL3442和BNL1064)在3年均被重复检测到,表型变异解释率最高分别为12.10%和8.02%。3个位点(Gh433-2、NAU3607和JESPR153)在2013,2014年同时检测到与黄萎病抗性显著关联;6个位点(BNL3902、NAU5428、BNL1646-1、BNL1646-2、CGR5996和HAU1774-1)在2014,2015年同时检测到与黄萎病抗性显著关联,其中,NAU5428的2年表型变异解释率较高,分别为6.93%和5.57%;其余16个位点仅在一个年份检测到与黄萎病抗性相关联。这些在不同年份中稳定存在的SSR标记位点有可能与抗病基因紧密连锁,有望用于棉花黄萎病抗性材料筛选和抗病基因挖掘中。

图4 SSR位点间的连锁不平衡
Fig.4 The linkage disequilibrium between SSR loci
表2 与黄萎病抗性相关的SSR位点
Tab.2 SSR loci associated with Verticillium wilt-resistance
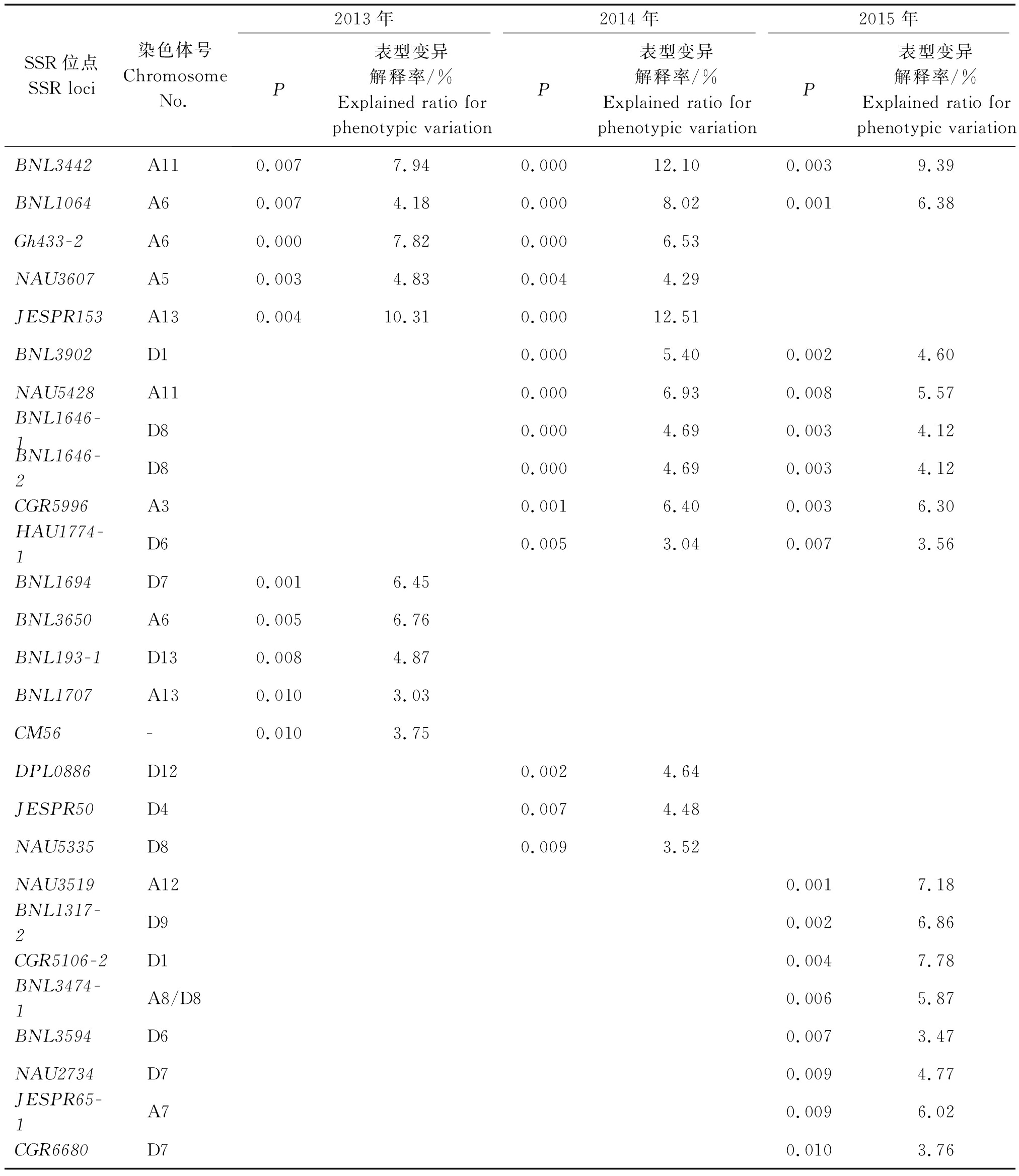
SSR位点SSR loci 染色体号Chromosome No.2013年2014年2015年P表型变异解释率/%Explained ratio for phenotypic variation P表型变异解释率/%Explained ratio for phenotypic variationP表型变异解释率/%Explained ratio for phenotypic variation BNL3442A110.007 7.94 0.000 12.10 0.003 9.39 BNL1064A60.007 4.18 0.000 8.02 0.001 6.38 Gh433-2A60.000 7.82 0.000 6.53 NAU3607A50.003 4.83 0.004 4.29 JESPR153A130.004 10.31 0.000 12.51 BNL3902D10.000 5.40 0.002 4.60 NAU5428A110.000 6.93 0.008 5.57 BNL1646-1D80.000 4.69 0.003 4.12 BNL1646-2D80.000 4.69 0.003 4.12 CGR5996A30.001 6.40 0.003 6.30 HAU1774-1D60.005 3.04 0.007 3.56 BNL1694D70.001 6.45 BNL3650A60.005 6.76 BNL193-1D130.008 4.87 BNL1707A130.010 3.03 CM56-0.010 3.75 DPL0886D120.002 4.64 JESPR50D40.007 4.48 NAU5335D80.009 3.52 NAU3519A120.001 7.18 BNL1317-2D90.002 6.86 CGR5106-2D10.004 7.78 BNL3474-1A8/D80.006 5.87 BNL3594D60.007 3.47 NAU2734D70.009 4.77 JESPR65-1A70.009 6.02 CGR6680D7 0.010 3.76
本研究通过对抗黄萎病性状进行关联分析的同时,挖掘出一些与棉花抗黄萎病稳定关联的SSR位点(BNL3442和BNL1064),可以为抗黄萎病候选位点做进一步验证,并挖掘优异等位变异,对棉花抗黄萎病分子育种具有重要的理论和实践意义。同时,本研究通过3年陆地棉资源的黄萎病抗性统计,鉴定了一批抗病性突出的育种材料和资源,为棉花抗病育种提供了参考。
目前,对于棉花黄萎病抗性的鉴定,主要有田间病圃鉴定、营养钵苗期鉴定和黄萎病毒素法鉴定[33]。其中,田间病圃鉴定是棉花种质抗黄萎病鉴定常用且能真实反映鉴定材料抗性强弱的方法,但由于大田鉴定存在不同材料成熟期早晚和接种量的差异,以及试验小区的干扰等,存在不同年份、不同棉区筛选的抗黄萎病材料的抗病性差异较大的情况。通过设置合适的感病对照品种,利用相对病情指数来描述棉花的抗病性是减少环境变异的有效措施[4,34]。本研究中,使用冀棉11同一批种子作为感病对照,最大程度保证了标准品种年度间抗病性的一致。本研究结果表明,尽管努力减少试验的环境误差,仍然存在部分材料在不同年份间鉴定结果差异较大的情况,可能是由于陆地棉黄萎病抗性属数量性状遗传[35],容易受到环境变化的影响。通过上述鉴定方法筛选出3年均表现为抗黄萎病的材料18份,这些材料中可能含有多环境稳定的抗黄萎病优势等位基因,需要在分子水平进行进一步挖掘。这些材料经其他鉴定方法重复检验后,有望作为抗黄萎病材料用于棉花育种工作中。
群体结构是影响关联分析的一个重要因素,研究一个群体的遗传结构组成时,会因为亚群的混合导致整个群体的连锁不平衡水平增强,假阳性结果增加[36],如果群体结构比较简单,假阳性关联结果出现的可能性就会减少[37]。因此,在进行关联分析前对自然群体进行遗传多样性和群体结构分析很有必要。本研究中,聚类结果显示,所选群体被分为3个亚群,群体结构分析表明,供试材料被分为2个亚群,2个聚类结果差异并不大,共同说明了所选陆地棉整体上遗传背景狭窄。这也解释了本研究中SSR引物在所选棉花品种中检测的等位变异数目和基因多样性的跨度较大,但平均值较低的问题。由于群体结构较为简单,本研究仅利用GLM模型进行了关联分析。
本研究通过关联分析获得了与黄萎病性状显著关联的SSR标记位点27个,与前人黄萎病抗性相关关联分析结果进行比较[18-20],Zhao等[20]利用212个SSR标记与陆地棉成株期和苗期的黄萎病抗性进行了关联分析,鉴定出NAU5428与黄萎病极显著相关(P<0.000 1),解释表型变异率达10.93%,本研究中,此标记在2年田间鉴定中与黄萎病显著相关,解释表型变异率分别为6.93%和5.57%,此结果表明了本研究结果与前人研究结果的一致性。
随着黄萎病抗性相关的QTLs的不断挖掘,已经有一些分子标记陆续应用于棉花抗病基因分子标记辅助选择育种上[4,38-41]。王芙蓉等[38]研究发现,标记 NAU751 和BNL1395 的抗性基因型聚合后可显著提高后代的抗性水平;孔祥瑞等[39]研究发现,BNL1721、BNL2733和 BNL3452这 3个标记的黄萎病抗性选择效应能够在不同世代间稳定遗传;祁伟彦等[40]利用和黄萎病抗性紧密连锁的 SSR 标记对棉花黄萎病抗性进行了辅助筛选,提出将人工病圃筛选和分子标记辅助育种相结合是选育棉花抗黄萎病材料可行、高效的育种方法。赵云雷等[4]研究表明,抗黄萎病优异等位基因的累加具有明显提高抗病性的作用,材料抗病性的强弱在一定程度上可以通过优异等位基因数目和效应值之和来反映,这就为分子方法鉴定抗病性强弱提供了依据,同时为黄萎病关联位点利用提供了重要参考。
通过关联分析挖掘了多个与陆地棉黄萎病抗性关联的分子标记位点,为棉花抗黄萎病性状的分子检测及标记辅助选择育种提供信息。今后应进一步挖掘与棉花抗黄萎病性状显著关联的优异等位基因位点,逐步实现抗病基因型的直接选择,有效缩短抗病性的鉴定周期。
[1] Fang L, Wang Q, Hu Y, et al.Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits[J]. Nature genetics, 2017, 49(7):1089-1098.
[2] Sal′kova E G, Guseva N N. The role of pectolytic enzymes of theVerticillium dahliae fungus in the development of cotton wilt[J]. Doklady Akademii Nauk SSR,1965, 163(2):515-522.
[3] Guo Xiuhua, Cai Caiping, Yuan Dongdong, et al. Development and identifi cation ofVerticillium wilt-resistant upland. cotton accessions by pyramiding QTL related to resistance[J]. Journal of Integrative Agriculture,2016, 15(3): 512-520.
[4] 赵云雷,王红梅,陈 伟,等. 基于优异等位基因的棉花抗黄萎病性状的分子鉴定[J]. 中国农业科学, 2017,50(2):216-227.
[5] Zhang Y, Wang X F, Ding Z G, et al.Transcriptome profiling ofGossypium barbadense inoculated withVerticillium dahliae provides a resource for cotton improvement[J]. BMC Genomics,2013, 14:637.
[6] Gapare W, Conaty W, Zhu Q H,et al. Genome-wide association study of yield components and fibre quality traits in a cotton germplasm diversity panel[J]. Euphytica, 2017, 213:66.
[7] Wang H M, Lin Z X, Zhang X L, et al. Mapping and quantitative trait loci analysis ofVerticillium wilt resistance genes in cotton[J]. Journal of Integrative Plant Biology,2008, 50(2):174-182.
[8] Jiang F, Zhao J, Zhou L, et al.Molecular mapping ofVerticillium wilt resistance QTL clustered on chromosomes D7 and D9 in upland cotton[J]. Science China, 2009,52:872-884.
[9] Fang H, Zhou H P, Sanogo S, et al. Quantitative trait locus mapping forVerticillium wilt resistance in a backcross inbred line population of cotton (Gossypium hirsutum Gossypium barbadense) based on RGA-AFLP analysis[J]. Euphytica, 2013, 194(1):79-91.
[10] Li C Q, Liu G S, Zhao H H, et al. Marker-assisted selection ofVerticillium wilt resistance in progeny populations of upland cotton derived from mass selection-mass crossing[J]. Euphytica,2013, 191(3):469-480.
[11] Ning Z Y, Zhao R, Chen H, et al. Molecular tagging of a major quantitative trait locus for broad-spectrum resistance toVerticillium wilt in upland cotton cultivar Prema[J]. Crop Science, 2013, 53(6):2304-2312.
[12] Zhang J F, Fang H, Zhou H P, et al. Genetics, breeding, and marker-assisted selection forVerticillium wilt resistance in cotton[J]. Crop Science, 2014,54:1289-1303.
[13] Zhang X, Yuan Y, Wei Z, et al. Molecular mapping and validation of a major QTL conferring resistance to a defoliating isolate ofVerticillium wilt in cotton (Gossypium hirsutum L.) [J]. PLoS One,2014,9(4):e96226.
[14] Flint-Garcia S A, Thuillet A C, Yu J M, et al. Maize association population: A high-resolution platform for quantitative trait locus dissection[J]. The Plant Journal, 2005, 44(6):1054-1064.
[15] Huang X, Han B. Natural variations and genome-wide association studies in crop plants[J]. Annu Rev Plant Biol,2014, 65:531-551.
[16] Nuzhdin S V, Friesen M L, McIntyre LM. Genotype-phenotype mapping in a post-GWAS world[J]. Trends Genet,2012, 28(9):421-426.
[17] Said J I, Song M Z, Wang H T, et al. Natural variations and genome-wide association studies in crop plants[J]. Annu Rev Plant Biol,2014, 65: 531-551.
[18] 郭志军,赵云雷,陈 伟,等. 陆地棉SSR标记遗传多样性及其与农艺性状的关联分析[J]. 棉花学报, 2014,26(5):420-430.
[19] 李黎贝. 陆地棉衰老和抗黄萎病相关性状的关联分析[D]. 安阳:中国农业科学院,2015.
[20] Zhao Y L, Wang H M, Chen Wei, et al. Genetic structure, linkage disequilibrium and association mapping ofVerticillium wilt resistance in elite cotton (Gossypium hirsutum L.) Germplasm Population[J]. PLoS One, 2014, 9(1):e86308. 66 ref.
[21] Su J, Fan S, Li L, et al. Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in Chinese upland cotton[J]. Frontiers in Plant Science, 2016, 7:1576.
[22] Islam M S, Thyssen G N, Jenkins J N, et al. A MAGIC population-based genome-wide association study reveals functional association ofGhRBB1_A07 gene with superior fiber quality in cotton[J]. BMC Genomics, 2016,17(1):903.
[23] Islam M S, Zeng L, Gregory N. Thyssen.: Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes[J]. Theor Appl Genet,2016b, 129(6):1071-1086.
[24] Sun Z, Wang X, Liu Z, et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits inGossypium hirsutum L.[J]. Plant Biotechnology Journal, 2017, 15(8):982-996.
[25] 朱荷琴,吴征彬,邹 奎,等. 国家棉花品种区域试验抗枯黄萎病鉴定方法[J]. 中国棉花,2007,34(11):9-24.
[26] Parerson A H,Brubaker C L,Wendel J F. A rapid method for extraction of cotton(Gossypium spp.) genomic DNA suitable for RFLP and PCR analysis[J]. Plant Mol Bio Rep,1993,11(2):122-127.
[27] Liang Zhao, Lü Yuanda, Cai Caiping, et al. Toward allotetraploid cotton genome assembly:integration of a high-density molecular genetic linkage map with DNA sequence information[J]. BMC Genomics, 2012, 13:539.
[28] 张 军,武耀廷,郭旺真,等. 棉花微卫星标记的PAGE/银染快速检测[J]. 棉花学报,2000,12(5):267-269.
[29] Liu K J, Muse S V. PowerMarker: an integrated analysis environment for genetic marker analysis[J]. Bioinformatics,2005, 21(9):2128-2129.
[30] Pritchard J K, Wen W. Documentation for STRUCTURE Software [M]. Version 2.2 Chicago: The University of Chicago Press, 2007.
[31] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE:a simulation study[J]. Molecular Ecology, 2005, 14:(8)2611-2620.
[32] Bradbury P J, Zhang Z, Kroon D E, et al. TASSEL:software for association mapping of complex traits in diverse samples [J]. Bioinformatics, 2007, 23(19):2633-2635.
[33] 房慧勇,马峙英.棉花抗黄萎病机制及抗病性鉴定研究进展[J]. 河北农业科学,2002,6(2):1-7.
[34] 李社增,马 平,Huang H C,等. 相对病情指数划分棉花品种杭病性的统计学基础[J]. 棉花学报,2003,15(6):344-347.
[35] 王红梅,张献龙,李运海,等. 陆地棉黄萎病抗性遗传分析[J]. 棉花学报,2004,16(2):84-88.
[36] 文自翔,赵团结,郑永战,等. 中国栽培和野生大豆农艺品质性状与SSR 标记的关联分析[J]. 作物学报, 2008,34(7):1169-1178.
[37] Wang Ju, Mc Clean P E, LeeR, et al. Association mapping of iron deficiency chlorosis loci in soybean(Glycine max L. Merr.)advanced breeding lines[J]. Theor Appl Genet, 2008, 116(6):777-787.
[38] 王芙蓉,刘任重,王留明,等. 陆地棉品种抗黄萎病性状的分子标记及其辅助选择效果[J].棉花学报, 2007, 19(6): 424-431.
[39] 孔祥瑞,王红梅,陈 伟,等.陆地棉黄萎病抗性的分子标记辅助选择效果[J].棉花学报,2010,22(6):527-532.
[40] 祁伟彦,张永军,张天真,等. 基于人工病圃筛选和分子标记辅助的棉花抗黄萎病育种方法研究与应用[J].分子植物育种,2012,10(5):607-612.
[41] 陈 红,李吉莲,刘 萍,等. 4个抗黄萎病海岛棉染色体片段导入系黄萎病抗性配合力分析[J]. 棉花学报,2014,26(4):290-294.