玉米叶夹角属于数量性状,受多基因控制。随着标记技术的发展,QTL定位方法大量应用于数量性状的研究。借助QTL作图,确定控制玉米叶夹角的基因位点,为玉米育种改良提供理论依据。Mickelson等[1]用玉米B73和Mo17 2个亲本构建重组自交系,采用符合区间作图法在2个环境中分别检测到7,3个控制玉米叶夹角的QTL;于永涛等[2]利用3个不同的F2:3群体玉米H21×Mo17、自330×K36和B73×L050,采用复合区间作图法检测到9个控制叶夹角的QTL;路明等[3]以掖478×丹340的F2为作图材料,采用复合区间作图法检测到6个与玉米叶夹角相关的QTL。但受到标记密度的限制,QTL检测的置信区间较大、有效性较低,难以为育种提供优良等位基因的信息。而全基因组关联分析(Genome-wide association study,GWAS)具有高分辨率、高通量的优势,有利于鉴定种质资源中的有利基因[4-11]。本研究利用全基因组关联分析结合玉米自然条件下的农艺性状对玉米穗上叶夹角进行初步定位,为进一步进行玉米叶夹角相关基因位点的精细定位、候选基因的功能及表达分析奠定理论基础。
1 材料和方法
1.1 试验材料
供试材料为289份玉米自交系构建的自然群体,包括我国骨干自交系及国外引进的优良自交系、山东省农业良种工程课题组选育的自交系。
1.2 田间试验设计
2014年在河北保定、山东枣庄,2015年在河北保定、河南洛阳种植289份玉米自交系,3 m行长,0.6 m行宽,每行15株,每个试验点3个重复。在玉米开花后15 d测量全部植株的叶夹角。
1.3 表型数据统计分析
利用SPSS 2.0软件对玉米叶夹角的表现型数据进行描述统计分析,获得不同环境下雄穗柄长的均值、方差、标准差、偏度、峰度及直方图。
1.4 玉米叶片总DNA的提取及基因型分析
参照CTAB法提取DNA[12],采用美国先锋公司开发的MaizeSNP50基因芯片进行基因分型。该芯片包括55 126个SNP标记,均匀分布在玉米B73的全基因组。参照Weng等[13]控制基因型数据质量。
1.5 SNP基因型的关联分析
从55 126个SNP中剔除缺失率大于20%、杂合率大于10%和最小基因频率低于0.05的标记,剩余25 331个基因型Marker,利用TASSEL 5.0软件对玉米雄穗柄长进行全基因组关联分析。
2 结果与分析
2.1 玉米叶夹角的表型数据分析
将已获得的玉米叶夹角表现型数据导入软件SPSS进行数据统计分析,同时得到不同环境下的直方图,如表1、图1所示。2014年保定、枣庄,2015年保定、洛阳4个环境玉米叶夹角的均值分别为26.25,29.32,27.57,24.41 cm,方差分别为56.477,81.562,54.015,28.254,标准差分别为7.515,9.031,7.350,5.315,如表1所示。4个环境下玉米叶夹角呈极显著相关。不同环境下玉米叶夹角的表现型数据虽然存在一定差异,但整体呈正态分布,植株具有一定的代表性,符合全基因组关联分析的要求,如图1所示。
表1 不同环境下玉米叶夹角的统计分析
Tab.1 Statistical analysis of leaf angle in maize under different environments
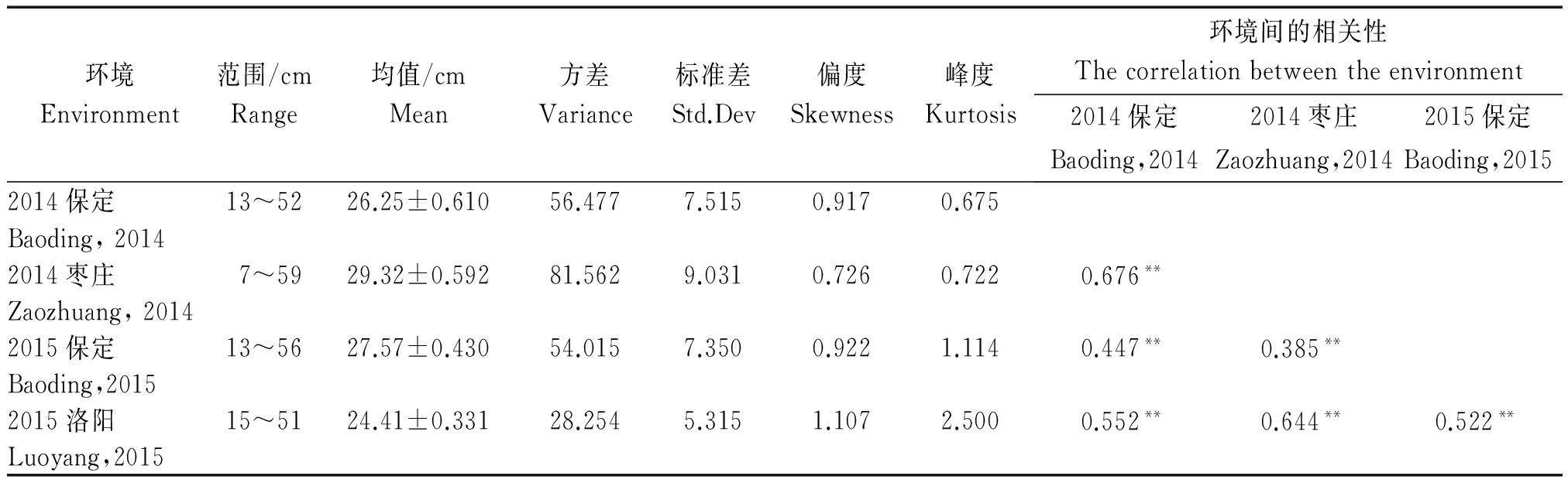
环境Environment范围/cmRange均值/cmMean方差Variance标准差Std.Dev偏度Skewness峰度Kurtosis环境间的相关性Thecorrelationbetweentheenvironment2014保定2014枣庄2015保定Baoding,2014Zaozhuang,2014Baoding,20152014保定13~5226.25±0.61056.4777.5150.9170.675Baoding,20142014枣庄7~5929.32±0.59281.5629.0310.7260.7220.676**Zaozhuang,20142015保定13~5627.57±0.43054.0157.3500.9221.1140.447**0.385**Baoding,20152015洛阳15~5124.41±0.33128.2545.3151.1072.5000.552**0.644**0.522**Luoyang,2015
2.2 玉米叶夹角的全基因组关联分析
将25 331个基因Marker与叶夹角用farmCPU进行全基因组关联分析。图2为玉米叶夹角的全基因组关联分析。图2-A、C、E、G为4个环境的曼哈顿图,纵轴间接表示各个标记与性状的关联性。图2-B、D、F、H为4个环境下玉米叶夹角全基因组关联分析的Q-Q图,横轴表示经过负的常数对数转换的期望P值,纵轴表示经过负的常数对数转换的观察到的P值。在2014年保定中筛选出14个标记,分布在1,2,3,4,5,8,9,10号染色体,如图2-A、表2。2014年枣庄筛选出10个标记,分布在1,5,7,8,9号染色体,如图2-C、表2。2015年保定筛选出10个标记,分布在1,5,6,7,8,10号染色体,如图2-E、表2。2015年洛阳筛选出8个标记,分布在1,3,5,8,9,10号染色体,如图2-G、表2。
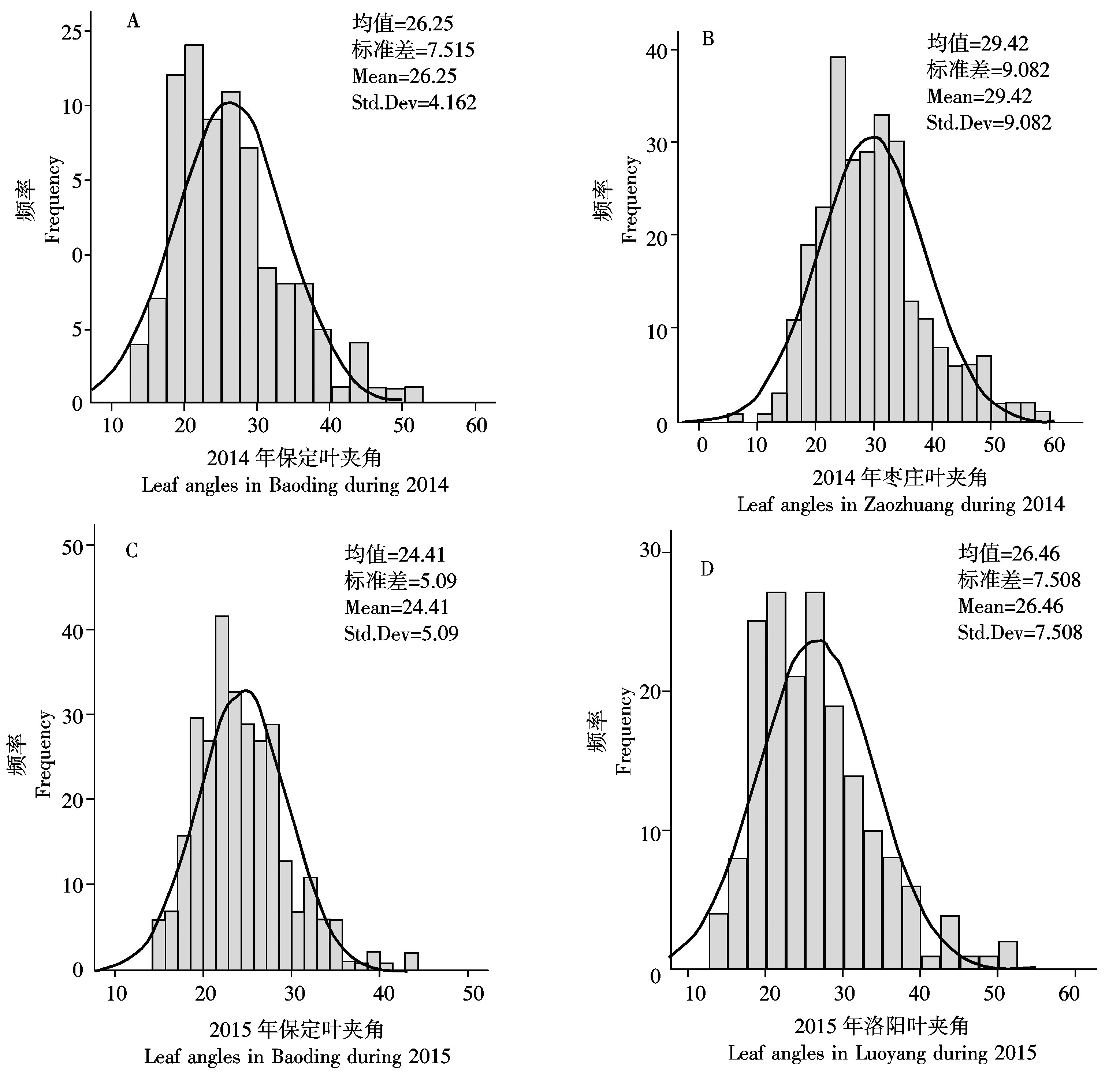
图1 玉米叶夹角表型数据的直方图
Fig.1 The histogram of leaf angles
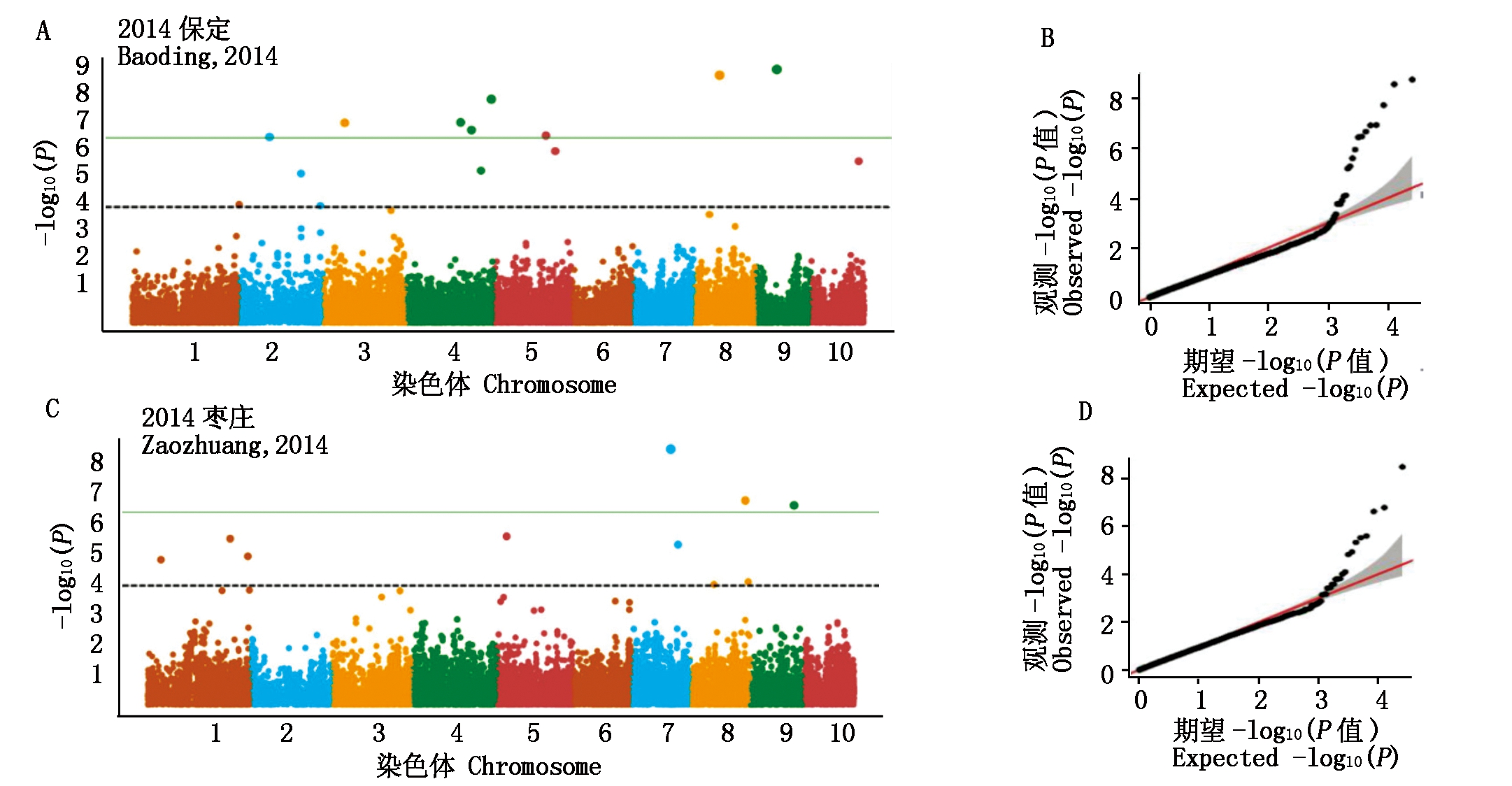
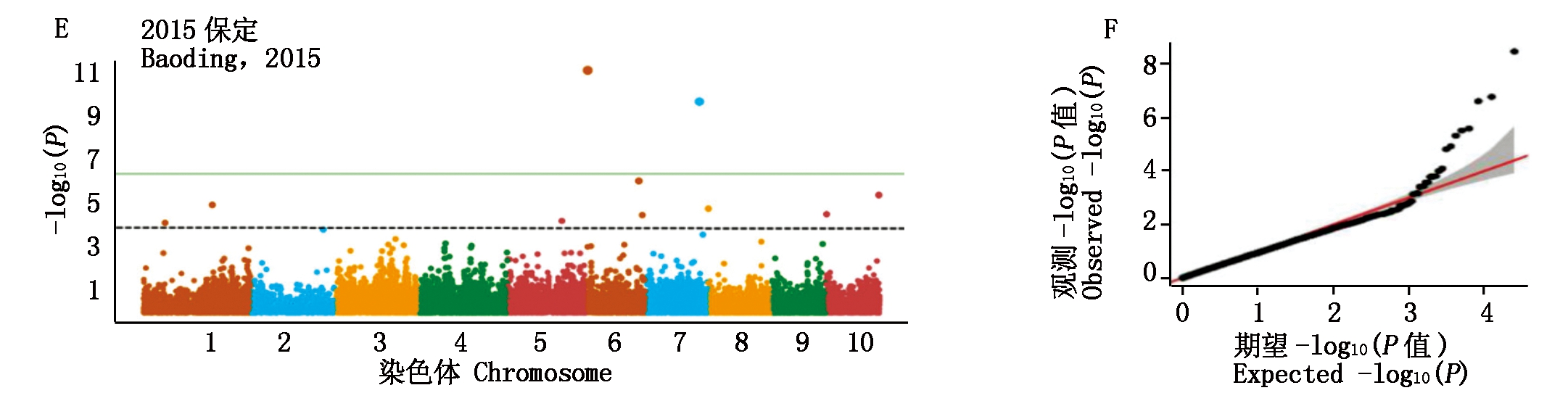
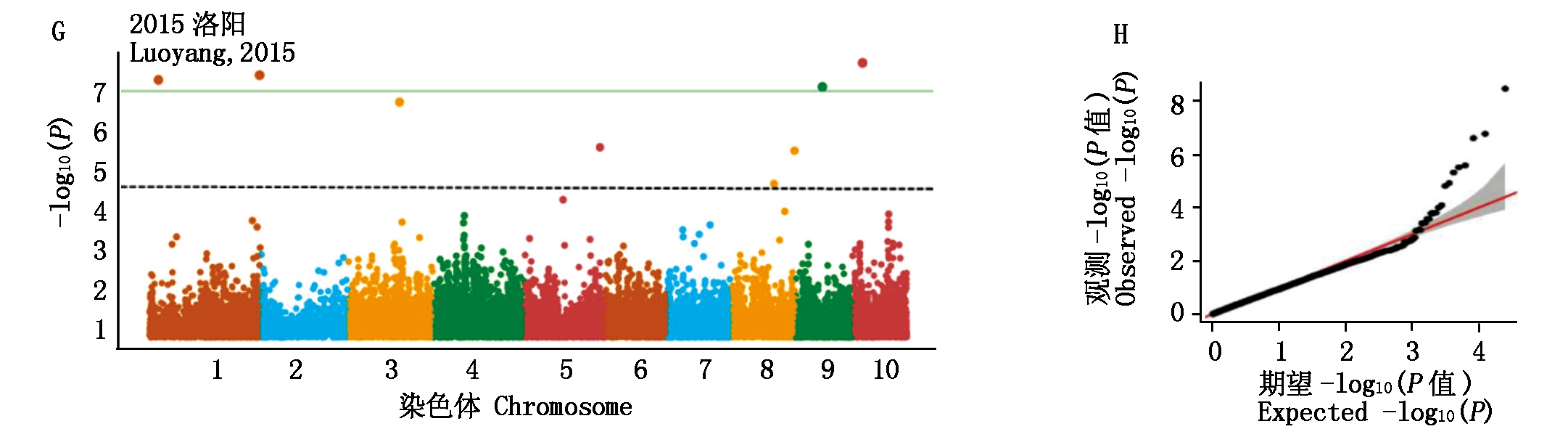
A.2014年保定叶夹角的曼哈顿图;B.2014年保定叶夹角全基因组关联分析的Q-Q图;C.2014年枣庄叶夹角的曼哈顿图;D.2014年枣庄叶夹角全基因组关联分析的Q-Q图;E.2015年保定叶夹角的曼哈顿图;F.2015年保定叶夹角全基因组关联分析的Q-Q图;G.2015年洛阳叶夹角的曼哈顿图;H.2015年洛阳叶夹角全基因组关联分析的Q-Q图。
A.Manhattan-map of leaf angle in Baoding during 2014;B.QQ-map of genome-wide association analysis of leaf angle in Baoding during 2014;C.Manhattan-map of leaf angle in Zaozhuang during 2014;D.QQ-map of genome-wide association analysis of leaf angle in Zaozhuang during 2014;E.Manhattan-map of leaf angle in Baoding during 2015;F.QQ-map of genome-wide association analysis of leaf angle in Baoding during 2015;G.Manhattan-map of leaf angle in Luoyang during 2015;H.QQ-map of genome-wide association analysis of leaf angle in Luoyang during 2015.
图2 玉米叶夹角全基因组关联分析
Fig.2 Genome-wide association analysis of leaf angle
表2 不同环境下各个染色体筛选的标记数
Tab.2 The number of markers selected for each chromosome in different environments
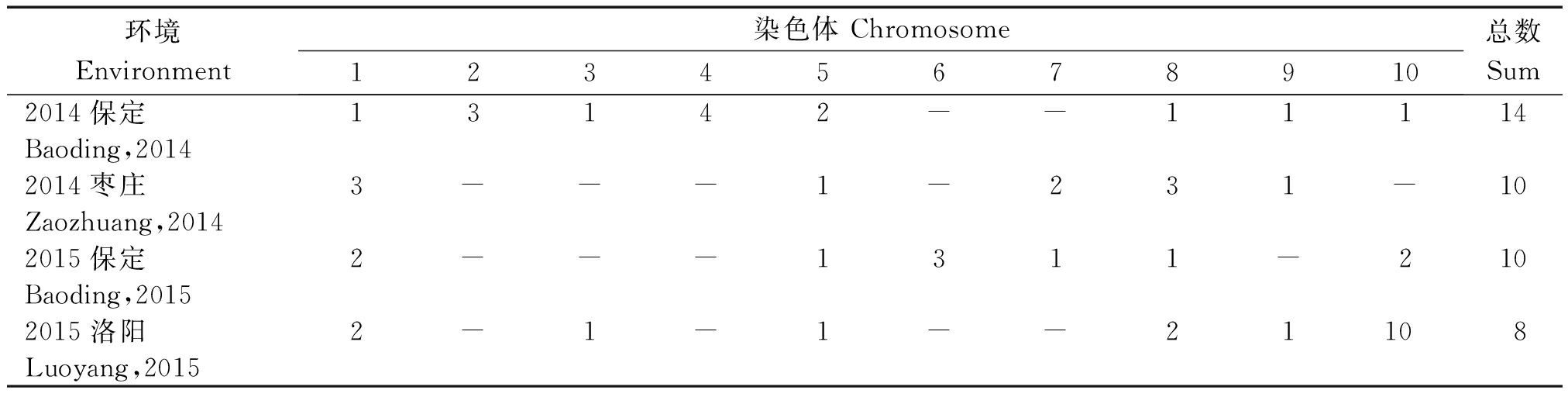
环境Environment染色体Chromosome12345678910总数Sum2014保定13142--11114Baoding,20142014枣庄3---1-231-10Zaozhuang,20142015保定2---1311-210Baoding,20152015洛阳2-1-1--21108Luoyang,2015
2.3 玉米叶夹角的候选基因分析
4个环境所筛选出的42个标记综合分析,共筛选出15个标记,分别位于Bin1.02、Bin1.03、Bin1.06、Bin1.11、Bin2.05、Bin3.04、Bin5.03、Bin5.04、Bin8.03、Bin8.04、Bin10.07处(表3)。
表3 15个与叶夹角显著相关的SNP位点
Tab.3 The 15 SNP associated with leaf angle

SNP基因型GenotypeBin物理位置Physicalposition候选基因CandidategeneP值P-value地点SiteSYN16268SYN34615SYN6426SYN38948SYN12784PZE_102088617PZE_103053199PZE_105040767PZE_105100624PZE_105098012PZE_105113489SYN12194PZE_108044479PZE_108066080PZE_110105428A/GC/TA/GG/TC/TC/TA/GA/GC/TC/TA/GA/GA/GC/TA/C1.021.031.061.111.112.053.045.035.045.045.048.038.038.0410.07 25425344398593731918902232992043502924264178842779060024683265847571508414651446130211704563647079545573420102117398624146554213pco120598si605022b11(700)LOC103642551LOC100191606LOC100279190LOC100284618LOC103627055LOC103628527LOC103627257umc1360ERD6IDP77LOC1036421662.03E-071.52E-051.04E-051.52E-071.16E-053.71E-071.21E-072.53E-065.57E-053.39E-071.14E-069.98E-052.71E-099.98E-053.64E-062015洛阳2014枣庄2015保定2015洛阳2014枣庄2014保定2014保定2014枣庄2015保定2014保定2014保定2014枣庄2014保定2015保定2015保定
3 讨论
GWAS是一种以连锁不平衡为基础,将SNP均匀分布于全基因组,借助统计学工具分析某一群体目标性状遗传变异的方法。目前,GWAS大量应用于鉴定植物病害、开花期、籽粒性状、株高等性状的研究[13-18]。本研究采用289份玉米自交系组成的关联作图群体对玉米雄穗柄长进行全基因组关联分析,在4个环境中共检测到42个与玉米叶夹角显著关联(P<0.000 01)的SNP。
Tuberosa等[19]提出,相同性状的QTL在不同环境下检测到,且效应方向相同,置信区间、标记区间重叠,可认为是同一QTL位点,Tian等[19-21]认为连锁定位和关联分析都可以检测数量性状位点,2种方法检测到的位点在位置上大部分具有一致性。结合前人的研究结果,本研究筛选出15个候选位点。Bin1.02、Bin1.06、Bin3.04、Bin10.07处筛选出与玉米穗上叶夹角相关的SNP标记与于永涛等[2]研究的结果在同一标记区间;Bin1.03处筛选的SNP标记位点与刘正等[22]的研究结果一致;Bin1.11处筛选出的SNP标记位点与刘兵[23]的研究结果一致;Bin2.05、Bin5.03、Bin8.03、Bin8.04处筛选出的SNP标记位点与刘鹏飞等[24]的研究结果处于同一区间。结果表明,以上区间极大可能存在与玉米叶夹角相关的基因位点。进一步研究时,可以在此区间适量加大标记密度。但本研究检测到的显著位点并非都与已经定位的QTL重叠,可能是与前人采用的双亲QTL作图法受亲本种质背景或检测微效QTL功效较低的影响有关。这表明采用全基因组关联分析策略是一种解析雄穗长遗传结构的有效方法[23,25]。
玉米叶夹角为多基因控制的数量性状,且受环境影响较大,研究过程比较复杂、可控性差,本试验结果可以为今后研究玉米叶夹角的基因位点提供参考依据,同时为进一步进行玉米叶夹角相关基因位点的精细定位、候选基因的功能及表达分析奠定理论基础,进而为玉米育种提供参考借鉴。
参考文献:
[1] Mickelson S M,Stuber C S,Senior L,et al.Quantitative trait loci controlling leaf and tassel traits in a B73×Mo17 population of maize[J].Crop Science,2002,42(6):1902-1909.
[2] 于永涛,张吉民,石云素,等.利用不同群体对玉米株高和叶片夹角的QTL分析[J].玉米科学,2006,14(2):88-92.
[3] 路 明,周 芳,谢传晓,等.玉米杂交种掖单13号的SSR连锁图谱构建与叶夹角和叶向值的QTL定位与分析[J].遗传,2007,29(9):1131-1138.
[4] Yu J,Buckler E S.Genetic association mapping and genome organization of maize[J].Current Opinion in Biotechnology,2006,17(2):155-160.
[5] Zhu Chengsong,Gore Michael,Buckler Edward S,et al.Status and prospects of association mapping in plants[J].The Plant Genome,2008,1(1):5-20.
[6] Brown P J,Upadyayula N,Mahone G S,et al.Distinct genetic architectures for male and female inflorescence traits of maize[J].PLoS Genetics,2011,7(11):e1002383.
[7] 许瀚元.基于SNP分子标记连锁图谱的玉米农艺性状QTL定位[D].扬州:扬州大学,2015.
[8] 贾 波,蒋思霞,邓德祥,等.玉米农艺性状QTL定位分析[J].玉米科学,2011,19(3):31-34.
[9] 高宝祯,刘 博,李石开,等.白菜类作物开花时间的全基因组关联分析[J].中国农业科学,2017,50(17):3375-3390.
[10] 熊姜玲,赵光伟,徐志红,等.甜瓜柄蔓夹角主基因+多基因遗传分析[J].中国农学通报,2016,32(34):55-61.
[11] 刘粉香,陶申童,吴吉妍,等.林木多元性状数据QTL区间作图统计分析及其在杨树上的应用[J].南京林业大学学报:自然科学版,2018(42):1-10.
[12] 高玉峰,张 攀,郝晓敏,等.一种快速提取玉米大群体基因组DNA的方法[J].中国农业大学学报,2011,16(6):32-36.
[13] Weng J F,Xie C X,Hao Z F,et al.Genome-Wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) inbred lines[J].PLoS One,2011,6(12):e29229.
[14] Yu J,Buckler E S.Genetic association mapping and genome organization of maize[J].Current Opinion in Biotechnology,2006,17(2):155-160.
[15] Buckler E S,Holland J B,Bradbury P J,et al.The genetic architecture of maize flowering time[J].Science,2009,325(5941):714-718.
[16] Kump K L,Bradbury P J,Wisser R J,et al.Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population[J].Nature Genetics,2011,43(2):163-168.
[17] 吴 律,代力强,董青松,等.玉米行粒数的全基因组关联分析[J].作物学报,2017,43(10):1559-1564.
[18] 张焕欣,翁建峰,张晓聪,等.玉米穗行数全基因组关联分析[J].作物学报,2014,40(1):1-6.
[19] Tuberosa R,Salvi S,Sanguineti M C,et al.Mapping QTLs regulating morpho-physiological traits and yield:case studies,shortcomings and perspectives in drought-stressed maize[J].Annals of Botany,2002,89 (7):941-963.
[20] 李永亮,王新宇,黄仕钰,等.大豆冠层参数的QTL定位及上位性互作分析[J].分子植物育种,2016(12):3414-3429.
[21] Tian F,Bradbury P J,Brown P J,et al.Genome-wide association study of leaf architecture in the maize nested association mapping population[J].Nature Genetics,2011,43(2):159-162.
[22] 刘 正,余婷婷,梅秀鹏,等.玉米穗上叶夹角和叶间距的QTL定位[J].农业生物技术学报,2014,22(2):177-187.
[23] 刘 兵.玉米叶夹角QTL定位[D].雅安:四川农业大学,2014.
[24] 刘鹏飞,蒋 锋,王汉宁,等.玉米叶夹角和叶向值的QTL定位[J].核农学报,2012,26(2):231-237.
[25] 张旷野,孙铭泽,闫 伟,等.玉米穗上叶叶夹角的遗传分析[J].作物杂志,2015(6):27-32.